publications
Publications categorized by year in reverse chronological order.
For the latest publications, visit Google Scholar.
2024
- BrJOphthalmolNovel loci for ocular axial length identified through extreme-phenotype genome-wide association study in Chinese populationsX. Han, S. Pan, J. Liu, and 11 more authorsBr J Ophthalmol, 2024
PURPOSE: To investigate genetic loci associated with ocular axial length (AL) in the Chinese population. METHODS: A genome-wide association study meta-analysis was conducted in totalling 2644 Chinese individuals from 3 cohorts: the Guangzhou cohort (GZ, 537 high myopes and 151 hyperopes), Wenzhou cohort (334 high myopes and 6 hyperopes) and Guangzhou Twin Eye Study (1051 participants with normally distributed AL). Functional mapping was performed to annotate the significant signals, possible tissues and cell types by integrating available multiomics data. Logistic regression models using AL-associated SNPs were constructed to predict three AL status in GZ. RESULTS: Two novel loci (1q25.2 FAM163A and 7p22.2 SDK1) showed genome-wide significant associations with AL, together explaining 29.63% of AL variance in GZ. The two lead SNPs improved the prediction accuracy for AL status, especially for hyperopes. The frequencies of AL decreasing (less myopic) alleles of the two SNPs were lowest in East Asians as compared with other populations (rs17370084: f (EAS)=0.03, f (EUR)=0.24, f (AFR)=0.05; rs73046501: f (EAS)=0.06, f (EUR)=0.07, f (AFR)=0.20), which was in line with the global distribution of myopia. The cerebral cortex and gamma-aminobutyric acidergic interneurons showed possible functional involvement in myopia development, and the galactose metabolic pathways were significantly enriched. CONCLUSION: Our study identified two population-specific novel loci for AL, expanding our understanding of the genetic basis of AL and providing evidence for a role of the nervous system and glucose metabolism in myopia pathogenesis.
- NatGenet
 Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traitsQ. Peng, X. Liu, W. Li, and 30 more authorsNat Genet, 2024
Analysis of blood methylation quantitative trait loci in East Asians reveals ancestry-specific impacts on complex traitsQ. Peng, X. Liu, W. Li, and 30 more authorsNat Genet, 2024Methylation quantitative trait loci (mQTLs) are essential for understanding the role of DNA methylation changes in genetic predisposition, yet they have not been fully characterized in East Asians (EAs). Here we identified mQTLs in whole blood from 3,523 Chinese individuals and replicated them in additional 1,858 Chinese individuals from two cohorts. Over 9% of mQTLs displayed specificity to EAs, facilitating the fine-mapping of EA-specific genetic associations, as shown for variants associated with height. Trans-mQTL hotspots revealed biological pathways contributing to EA-specific genetic associations, including an ERG-mediated 233 trans-mCpG network, implicated in hematopoietic cell differentiation, which likely reflects binding efficiency modulation of the ERG protein complex. More than 90% of mQTLs were shared between different blood cell lineages, with a smaller fraction of lineage-specific mQTLs displaying preferential hypomethylation in the respective lineages. Our study provides new insights into the mQTL landscape across genetic ancestries and their downstream effects on cellular processes and diseases/traits.
- IJLMA methylation panel of 10 CpGs for accurate age inference via stepwise conditional epigenome-wide association studyY. Qian, Q. Peng, Q. Qian, and 10 more authorsInt J Legal Med, 2024
Estimating individual age from DNA methylation at age associated CpG sites may provide key information facilitating forensic investigations. Systematic marker screening and feature selection play a critical role in ensuring the performance of the final prediction model. In the discovery stage, we screened for 811876 CpGs from whole blood of 2664 Chinese individuals ranging from 18 to 83 years of age based on a stepwise conditional epigenome-wide association study (SCEWAS). The SCEWAS identified 28 CpGs showing genome-wide significant and independent effects. Further restricting this panel to 10 most informative CpGs showed a tolerable loss of information. A linear model consisting of these 10 CpGs could explain 93% of the age variance (R(2) = 0.93) in the training set (n = 2664). In an independent test set of Chinese individuals (n = 648), this model also provided highly accurate predictions (R(2) = 0.85, mean absolute deviation, MAD = 3.20 years). The model was additionally validated in a public dataset of multiple ancestral origins (86 Europeans, 14 Asians, and 273 Africans) and the prediction accuracy reduced significantly (R(2) = 0.85, MAD = 6.21 years), as might be expected due to different genomic backgrounds, sample sizes, and age ranges. Our 10 CpG model also outperformed the recently proposed 9-CpG model constructed in 390 Chinese males (R(2) = 0.79 in test set). We also demonstrated that our SCEWAS approach outperformed the traditional EWAS and the elastic net approach in obtaining a small set of most age informative CpGs. Overall, our systematic genome-wide feature selection identified a small panel of 10 CpGs for accurate age estimation with high potential in forensic applications.
- JEADVDeep learning predicted perceived age is a reliable approach for analysis of facial ageing: A proof of principle studyC. Turner, L. M. Pardo, D. A. Gunn, and 14 more authorsJ Eur Acad Dermatol Venereol, 2024
BACKGROUND: Perceived age (PA) has been associated with mortality, genetic variants linked to ageing and several age-related morbidities. However, estimating PA in large datasets is laborious and costly to generate, limiting its practical applicability. OBJECTIVES: To determine if estimating PA using deep learning-based algorithms results in the same associations with morbidities and genetic variants as human-estimated perceived age. METHODS: Self-supervised learning (SSL) and deep feature transfer (DFT) deep learning (DL) approaches were trained and tested on human-estimated PAs and their corresponding frontal face images of middle-aged to elderly Dutch participants (n = 2679) from a population-based study in the Netherlands. We compared the DL-estimated PAs with morbidities previously associated with human-estimated PA as well as genetic variants in the gene MC1R; we additionally tested the PA associations with MC1R in a new validation cohort (n = 1158). RESULTS: The DL approaches predicted PA in this population with a mean absolute error of 2.84 years (DFT) and 2.39 years (SSL). In the training-test dataset, we found the same significant (p < 0.05) associations for DL PA with osteoporosis, ARHL, cognition, COPD and cataracts and MC1R, as with human PA. We also found a similar but less significant association for SSL and DFT PAs (0.69 and 0.71 years per allele, p = 0.008 and 0.011, respectively) with MC1R variants in the validation dataset as that found with human, SSL and DFT PAs in the training-test dataset (0.79, 0.78 and 0.71 years per allele respectively; all p < 0.0001). CONCLUSIONS: Deep learning methods can automatically estimate PA from facial images with enough accuracy to replicate known links between human-estimated perceived age and several age-related morbidities. Furthermore, DL predicted perceived age associated with MC1R gene variants in a validation cohort. Hence, such DL PA techniques may be used instead of human estimations in perceived age studies thereby reducing time and costs.
- PLoSGenetSearching across-cohort relatives in 54,092 GWAS samples via encrypted genotype regressionQ. X. Zhang, T. Liu, X. Guo, and 22 more authorsPLoS Genet, 2024
Explicitly sharing individual level data in genomics studies has many merits comparing to sharing summary statistics, including more strict QCs, common statistical analyses, relative identification and improved statistical power in GWAS, but it is hampered by privacy or ethical constraints. In this study, we developed encG-reg, a regression approach that can detect relatives of various degrees based on encrypted genomic data, which is immune of ethical constraints. The encryption properties of encG-reg are based on the random matrix theory by masking the original genotypic matrix without sacrificing precision of individual-level genotype data. We established a connection between the dimension of a random matrix, which masked genotype matrices, and the required precision of a study for encrypted genotype data. encG-reg has false positive and false negative rates equivalent to sharing original individual level data, and is computationally efficient when searching relatives. We split the UK Biobank into their respective centers, and then encrypted the genotype data. We observed that the relatives estimated using encG-reg was equivalently accurate with the estimation by KING, which is a widely used software but requires original genotype data. In a more complex application, we launched a finely devised multi-center collaboration across 5 research institutes in China, covering 9 cohorts of 54,092 GWAS samples. encG-reg again identified true relatives existing across the cohorts with even different ethnic backgrounds and genotypic qualities. Our study clearly demonstrates that encrypted genomic data can be used for data sharing without loss of information or data sharing barrier.

2023
- FSIAEAn announcement of a new genome sequence available for Dama dama (fallow deer)Rebecca K. Barnard, Judith A. Smith, Na Yuan, and 2 more authorsForensic Science International: Animals and Environments, 2023
- EJHGGenetic prediction of male pattern baldness based on large independent datasetsY. Chen, P. Hysi, C. Maj, and 4 more authorsEur J Hum Genet, 2023
Genetic prediction of male pattern baldness (MPB) is important in science and society. Previous genetic MPB prediction models were limited by sparse marker coverage, small sample size, and/or data dependency in the different analytical steps. Here, we present novel models for genetic prediction of MPB based on a large set of markers and large independent subsample sets drawn among 187,435 European subjects. We selected 117 SNP predictors within 85 distinct loci from a list of 270 previously MPB-associated SNPs in 55,573 males of the UK Biobank Study (UKBB). Based on these 117 SNPs with and without age as additional predictor, we trained, by use of different methods, prediction models in a non-overlapping subset of 104,694 UKBB males and tested them in a non-overlapping subset of 26,177 UKBB males. Estimates of prediction accuracy were similar between methods with AUC ranges of 0.725-0.728 for severe, 0.631-0.635 for moderate, 0.598-0.602 for slight, and 0.708-0.711 for no hair loss with age, and slightly lower without, while prediction of any versus no hair loss gave 0.690-0.711 with age and slightly lower without. External validation in an early-onset enriched MPB dataset from the Bonn Study (N = 991) showed improved prediction accuracy without considering age such as AUC of 0.830 for no vs. any hair loss. Because of the large number of markers and the large independent datasets used for the different analytical steps, the newly presented genetic prediction models are the most reliable ones currently available for MPB or any other human appearance trait.
- FSMPEvaluation of facial hair-associated SNPs: a pilot study on male Pakistani Punjabi populationM. Jawad, A. Adnan, R. A. Rehman, and 7 more authorsForensic Sci Med Pathol, 2023
Variation in facial hair is one of the most conspicuous features of facial appearance, particularly in South Asia and Middle East countries. A genome-wide association study in Latin Americans has identified multiple genetic variants at distinct loci being associated with facial hair traits including eyebrow thickness, beard thickness, and monobrow. In this pilot study, we have evaluated 16 SNPs associated with facial hair traits in 58 male individuals from the Punjabi population of Pakistan. In our sample, rs365060 in EDAR and rs12597422 in FTO showed significant association with monobrow, rs6684877 in MACF1 showed significant association with eyebrow thickness, and two SNPs in LOC105379031 (rs9654415 and rs7702331) showed significant association with beard thickness. Our results also suggest that genetic association may vary between ethnic groups and geographic regions. Although more data are needed to validate our results, our findings are of value in forensic molecular photofitting research in Pakistan.
- PLoSGenetCombined genome-wide association study of 136 quantitative ear morphology traits in multiple populations reveal 8 novel lociY. Li, Z. Xiong, M. Zhang, and 31 more authorsPLoS Genet, 2023
Human ear morphology, a complex anatomical structure represented by a multidimensional set of correlated and heritable phenotypes, has a poorly understood genetic architecture. In this study, we quantitatively assessed 136 ear morphology traits using deep learning analysis of digital face images in 14,921 individuals from five different cohorts in Europe, Asia, and Latin America. Through GWAS meta-analysis and C-GWASs, a recently introduced method to effectively combine GWASs of many traits, we identified 16 genetic loci involved in various ear phenotypes, eight of which have not been previously associated with human ear features. Our findings suggest that ear morphology shares genetic determinants with other surface ectoderm-derived traits such as facial variation, mono eyebrow, and male pattern baldness. Our results enhance the genetic understanding of human ear morphology and shed light on the shared genetic contributors of different surface ectoderm-derived phenotypes. Additionally, gene editing experiments in mice have demonstrated that knocking out the newly ear-associated gene (Intu) and a previously ear-associated gene (Tbx15) causes deviating mouse ear morphology.
- Eur J EpidemiolGenome-wide epistasis study highlights genetic interactions influencing severity of COVID-19S. Lin, X. Gao, F. Degenhardt, and 14 more authorsEur J Epidemiol, 2023
Coronavirus disease 2019 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may lead to life-threatening respiratory symptoms. Understanding the genetic basis of the prognosis of COVID-19 is important for risk profiling of potentially severe symptoms. Here, we conducted a genome-wide epistasis study of COVID-19 severity in 2243 patients with severe symptoms and 12,612 patients with no or mild symptoms from the UK Biobank, followed by a replication study in an independent Spanish cohort (1416 cases, 4382 controls). Our study highlighted 3 interactions with genome-wide significance in the discovery phase, nominally significant in the replication phase, and enhanced significance in the meta-analysis. For example, the lead interaction was found between rs9792388 upstream of PDGFRL and rs3025892 downstream of SNAP25, where the composite genotype of rs3025892 CT and rs9792388 CA/AA showed higher risk of severe disease than any other genotypes (P = 2.77 x 10(-12), proportion of severe cases = 0.24 0.29 vs. 0.09 0.18, genotypic OR = 1.96 2.70). This interaction was replicated in the Spanish cohort (P = 0.002, proportion of severe cases = 0.30 0.36 vs. 0.14 0.25, genotypic OR = 1.45 2.37) and showed enhanced significance in the meta-analysis (P = 4.97 x 10(-14)). Notably, these interactions indicated a possible molecular mechanism by which SARS-CoV-2 affects the nervous system. The first exhaustive genome-wide screening for epistasis improved our understanding of genetic basis underlying the severity of COVID-19.
- FrontImmunolT cell receptor beta repertoires in patients with COVID-19 reveal disease severity signaturesJ. Xu, X. X. Li, N. Yuan, and 16 more authorsFront Immunol, 2023
BACKGROUND: The immune responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are crucial in maintaining a delicate balance between protective effects and harmful pathological reactions that drive the progression of coronavirus disease 2019 (COVID-19). T cells play a significant role in adaptive antiviral immune responses, making it valuable to investigate the heterogeneity and diversity of SARS-CoV-2-specific T cell responses in COVID-19 patients with varying disease severity. METHODS: In this study, we employed high-throughput T cell receptor (TCR) beta repertoire sequencing to analyze TCR profiles in the peripheral blood of 192 patients with COVID-19, including those with moderate, severe, or critical symptoms, and compared them with 81 healthy controls. We specifically focused on SARS-CoV-2-associated TCR clonotypes. RESULTS: We observed a decrease in the diversity of TCR clonotypes in COVID-19 patients compared to healthy controls. However, the overall abundance of dominant clones increased with disease severity. Additionally, we identified significant differences in the genomic rearrangement of variable (V), joining (J), and VJ pairings between the patient groups. Furthermore, the SARS-CoV-2-associated TCRs we identified enabled accurate differentiation between COVID-19 patients and healthy controls (AUC > 0.98) and distinguished those with moderate symptoms from those with more severe forms of the disease (AUC > 0.8). These findings suggest that TCR repertoires can serve as informative biomarkers for monitoring COVID-19 progression. CONCLUSIONS: Our study provides valuable insights into TCR repertoire signatures that can be utilized to assess host immunity to COVID-19. These findings have important implications for the use of TCR beta repertoires in monitoring disease development and indicating disease severity.
2022
- JGGIdentification of novel loci influencing refractive error in East Asian populations using an extreme phenotype designX. Han, T. Liu, X. Ding, and 17 more authorsJ Genet Genomics, 2022
The global "myopia boom" has raised significant international concerns. Despite a higher myopia prevalence in Asia, previous large-scale genome-wide association studies (GWASs) were mostly based on European descendants. Here, we report a GWAS of spherical equivalent (SE) in 1852 Chinese Han individuals with extreme SE from Guangzhou (631 < -6.00D and 574 > 0.00D) and Wenzhou (593 < -6.00D and 54 > -1.75D), followed by a replication study in two independent cohorts with totaling 3538 East Asian individuals. The discovery GWAS and meta-analysis identify three novel loci, which show genome-wide significant associations with SE, including 1q25.2 FAM163A, 10p11.22 NRP1/PRAD3, and 10p11.21 ANKRD30A/MTRNR2L7, together explaining 3.34% of SE variance. 10p11.21 is successfully replicated. The allele frequencies of all three loci show significant differences between major continental groups (P < 0.001). The SE reducing (more myopic) allele of rs10913877 (1q25.2 FAM163A) demonstrates the highest frequency in East Asians and much lower frequencies in Europeans and Africans (EAS = 0.60, EUR = 0.20, and AFR = 0.18). The gene-based analysis additionally identifies three novel genes associated with SE, including EI24, LHX5, and ARPP19. These results provide new insights into myopia pathogenesis and indicate the role of genetic heterogeneity in myopia epidemiology among different ethnicities.
- FrontGenetCausal Inference of Genetic Variants and Genes in Amyotrophic Lateral SclerosisS. Pan, X. Liu, T. Liu, and 5 more authorsFront Genet, 2022
Amyotrophic lateral sclerosis (ALS) is a fatal progressive multisystem disorder with limited therapeutic options. Although genome-wide association studies (GWASs) have revealed multiple ALS susceptibility loci, the exact identities of causal variants, genes, cell types, tissues, and their functional roles in the development of ALS remain largely unknown. Here, we reported a comprehensive post-GWAS analysis of the recent large ALS GWAS (n = 80,610), including functional mapping and annotation (FUMA), transcriptome-wide association study (TWAS), colocalization (COLOC), and summary data-based Mendelian randomization analyses (SMR) in extensive multi-omics datasets. Gene property analysis highlighted inhibitory neuron 6, oligodendrocytes, and GABAergic neurons (Gad1/Gad2) as functional cell types of ALS and confirmed cerebellum and cerebellar hemisphere as functional tissues of ALS. Functional annotation detected the presence of multiple deleterious variants at three loci (9p21.2, 12q13.3, and 12q14.2) and highlighted a list of SNPs that are potentially functional. TWAS, COLOC, and SMR identified 43 genes at 24 loci, including 23 novel genes and 10 novel loci, showing significant evidence of causality. Integrating multiple lines of evidence, we further proposed that rs2453555 at 9p21.2 and rs229243 at 14q12 functionally contribute to the development of ALS by regulating the expression of C9orf72 in pituitary and SCFD1 in skeletal muscle, respectively. Together, these results advance our understanding of the biological etiology of ALS, feed into new therapies, and provide a guide for subsequent functional experiments.
- JGGGenetic evidence for facial variation being a composite phenotype of cranial variation and facial soft tissue thicknessW. Qian, M. Zhang, K. Wan, and 10 more authorsJ Genet Genomics, 2022
Facial and cranial variation represent a multidimensional set of highly correlated and heritable phenotypes. Little is known about the genetic basis explaining this correlation. We develop a software package ALoSFL for simultaneous localization of facial and cranial landmarks from head computed tomography (CT) images, apply it in the analysis of head CT images of 777 Han Chinese women, and obtain a set of phenotypes representing variation in face, skull and facial soft tissue thickness (FSTT). Association analysis of 301 single nucleotide polymorphisms (SNPs) from 191 distinct genomic loci previously associated with facial variation reveals an unexpected larger number of loci showing significant associations (P < 1e-3) with cranial phenotypes than expected under the null (O/E = 3.39), suggesting facial and cranial phenotypes share a substantial proportion of genetic components. Adding FSTT to a SNP-only model shows a large impact in explaining facial variance. A gene ontology analysis reveals that bone morphogenesis and osteoblast differentiation likely underlie our cranial-significant findings. Overall, this study simultaneously investigates the genetic effects on both facial and cranial variation of the same sample, supporting that facial variation is a composite phenotype of cranial variation and FSTT.
- NatCommun
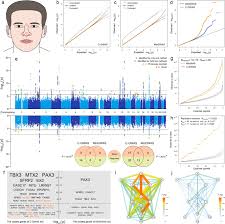 Combining genome-wide association studies highlight novel loci involved in human facial variationZ. Xiong, X. Gao, Y. Chen, and 11 more authorsNat Commun, 2022
Combining genome-wide association studies highlight novel loci involved in human facial variationZ. Xiong, X. Gao, Y. Chen, and 11 more authorsNat Commun, 2022Standard genome-wide association studies (GWASs) rely on analyzing a single trait at a time. However, many human phenotypes are complex and composed by multiple correlated traits. Here we introduce C-GWAS, a method for combining GWAS summary statistics of multiple potentially correlated traits. Extensive computer simulations demonstrated increased statistical power of C-GWAS compared to the minimal p-values of multiple single-trait GWASs (MinGWAS) and the current state-of-the-art method for combining single-trait GWASs (MTAG). Applying C-GWAS to a meta-analysis dataset of 78 single trait facial GWASs from 10,115 Europeans identified 56 study-wide suggestively significant loci with multi-trait effects on facial morphology of which 17 are novel loci. Using data from additional 13,622 European and Asian samples, 46 (82%) loci, including 9 (53%) novel loci, were replicated at nominal significance with consistent allele effects. Functional analyses further strengthen the reliability of our C-GWAS findings. Our study introduces the C-GWAS method and makes it available as computationally efficient open-source R package for widespread future use. Our work also provides insights into the genetic architecture of human facial appearance.

2021
- SciAdvA GWAS in Latin Americans identifies novel face shape loci, implicating VPS13B and a Denisovan introgressed region in facial variationB. Bonfante, P. Faux, N. Navarro, and 51 more authorsSci Adv, 2021
To characterize the genetic basis of facial features in Latin Americans, we performed a genome-wide association study (GWAS) of more than 6000 individuals using 59 landmark-based measurements from two-dimensional profile photographs and 9,000,000 genotyped or imputed single-nucleotide polymorphisms. We detected significant association of 32 traits with at least 1 (and up to 6) of 32 different genomic regions, more than doubling the number of robustly associated face morphology loci reported until now (from 11 to 23). These GWAS hits are strongly enriched in regulatory sequences active specifically during craniofacial development. The associated region in 1p12 includes a tract of archaic adaptive introgression, with a Denisovan haplotype common in Native Americans affecting particularly lip thickness. Among the nine previously unidentified face morphology loci we identified is the VPS13B gene region, and we show that variants in this region also affect midfacial morphology in mice.
- FSIGThe impact of correlations between pigmentation phenotypes and underlying genotypes on genetic prediction of pigmentation traitsY. Chen, W. Branicki, S. Walsh, and 4 more authorsForensic Sci Int Genet, 2021
Predicting appearance phenotypes from genotypes is relevant for various areas of human genetic research and applications such as genetic epidemiology, human history, anthropology, and particularly in forensics. Many appearance phenotypes, and thus their underlying genotypes, are highly correlated, with pigmentation traits serving as primary examples. However, all available genetic prediction models, including those for pigmentation traits currently used in forensic DNA phenotyping, ignore phenotype correlations. Here, we investigated the impact of appearance phenotype correlations on genetic appearance prediction in the exemplary case of three pigmentation traits. We used data for categorical eye, hair and skin colour as well as 41 DNA markers utilized in the recently established HIrisPlex-S system from 762 individuals with complete phenotype and genotype information. Based on these data, we performed genetic prediction modelling of eye, hair and skin colour via three different strategies, namely the established approach of predicting phenotypes solely based on genotypes while not considering phenotype correlations, and two novel approaches that considered phenotype correlations, either incorporating truly observed correlated phenotypes or DNA-predicted correlated phenotypes in addition to the DNA predictors. We found that using truly observed correlated pigmentation phenotypes as additional predictors increased the DNA-based prediction accuracies for almost all eye, hair and skin colour categories, with the largest increase for intermediate eye colour, brown hair colour, dark to black skin colour, and particularly for dark skin colour. Outcomes of dedicated computer simulations suggest that this prediction accuracy increase is due to the additional genetic information that is implicitly provided by the truly observed correlated pigmentation phenotypes used, yet not covered by the DNA predictors applied. In contrast, considering DNA-predicted correlated pigmentation phenotypes as additional predictors did not improve the performance of the genetic prediction of eye, hair and skin colour, which was in line with the results from our computer simulations. Hence, in practical applications of DNA-based appearance prediction where no phenotype knowledge is available, such as in forensic DNA phenotyping, it is not advised to use DNA-predicted correlated phenotypes as predictors in addition to the DNA predictors. In the very least, this is not recommended for the pigmentation traits and the established pigmentation DNA predictors tested here.
- CommunBiolhReg-CNCC reconstructs a regulatory network in human cranial neural crest cells and annotates variants in a developmental contextZ. Feng, Z. Duren, Z. Xiong, and 4 more authorsCommun Biol, 2021
Cranial Neural Crest Cells (CNCC) originate at the cephalic region from forebrain, midbrain and hindbrain, migrate into the developing craniofacial region, and subsequently differentiate into multiple cell types. The entire specification, delamination, migration, and differentiation process is highly regulated and abnormalities during this craniofacial development cause birth defects. To better understand the molecular networks underlying CNCC, we integrate paired gene expression & chromatin accessibility data and reconstruct the genome-wide human Regulatory network of CNCC (hReg-CNCC). Consensus optimization predicts high-quality regulations and reveals the architecture of upstream, core, and downstream transcription factors that are associated with functions of neural plate border, specification, and migration. hReg-CNCC allows us to annotate genetic variants of human facial GWAS and disease traits with associated cis-regulatory modules, transcription factors, and target genes. For example, we reveal the distal and combinatorial regulation of multiple SNPs to core TF ALX1 and associations to facial distances and cranial rare disease. In addition, hReg-CNCC connects the DNA sequence differences in evolution, such as ultra-conserved elements and human accelerated regions, with gene expression and phenotype. hReg-CNCC provides a valuable resource to interpret genetic variants as early as gastrulation during embryonic development. The network resources are available at https://github.com/AMSSwanglab/hReg-CNCC .
- FrontGenetExome-Wide Association Study Identifies East Asian-Specific Missense Variant MTHFR C136T Influencing Homocysteine Levels in Chinese Populations RH: ExWAS of tHCY in a Chinese PopulationT. Liu, M. Momin, H. Zhou, and 15 more authorsFront Genet, 2021
Plasma total homocysteine (tHCY) is a known risk factor of a wide range of complex diseases. No genome scans for tHCY have been conducted in East Asian populations. Here, we conducted an exome-wide association study (ExWAS) for tHCY in 5,175 individuals of Chinese Han origin, followed by a replication study in 668 Chinese individuals. The ExWAS identified two loci, 1p36.22 (lead single-nucleotide polymorphism (SNP) rs1801133, MTHFR C677T) and 16q24.3 (rs1126464, DPEP1), showing exome-wide significant association with tHCY (p < 5E-7); and both loci have been previously associated with tHCY in non-East Asian populations. Both SNPs were replicated in the replication study (p < 0.05). Conditioning on the genotype of C677T and rs1126464, we identified a novel East Asian-specific missense variant rs138189536 (C136T) of MTHFR (p = 6.53E-10), which was also significant in the replication study (p = 9.8E-3). The C136T and C677T variants affect tHCY in a compound heterozygote manner, where compound heterozygote and homozygote genotype carriers had on average 43.4% increased tHCY than had other genotypes. The frequency of the homozygote C677T genotype showed an inverse-U-shaped geospatial pattern globally with a pronounced frequency in northern China, which coincided with the high prevalence of hyperhomocysteinemia (HHCY) in northern China. A logistic regression model of HHCY status considering sex, age, and the genotypes of the three identified variants reached an area under the receiver operating characteristic curve (AUC) value of 0.74 in an independent validation cohort. These genetic observations provide new insights into the presence of multiple causal mutations at the MTHFR locus, highlight the role of genetics in HHCY epidemiology among different populations, and provide candidate loci for future functional studies.
- FASEBJEvidence for CAT gene being functionally involved in the susceptibility of COVID-19Y. Qian, Y. Li, X. Liu, and 4 more authorsFASEB J, 2021
Novel coronary pneumonia (COVID-19) is a respiratory distress syndrome caused by a new type of coronavirus. Understanding the genetic basis of susceptibility and prognosis to COVID-19 is of great significance to disease prevention, molecular typing, prognosis, and treatment. However, so far, there have been only two genome-wide association studies (GWASs) on the susceptibility of COVID-19. Starting with these reported DNA variants, we found the genes regulated by these variants through cis-eQTL and cis-meQTL acting. We further did a series of bioinformatics analysis on these potential risk genes. The analysis shows that the genetic variants on EHF regulate the expression of its neighbor CAT gene via cis-eQTL. There was significant evidence that CAT and the SARS-CoV-2-related S protein binding protein ACE2 interact with each other. Intracellular localization results showed that CAT and ACE2 proteins both exists in the cell membrane and extracellular area and their interaction could have an impact on the cell invasion ability of S protein. In addition, the expression of these three genes showed a significant positive correlation in the lungs. Based on these results, we propose that CAT plays a crucial intermediary role in binding effectiveness of ACE2, thereby affecting the susceptibility to COVID-19.
- FASEBBioadvThe effects of Tbx15 and Pax1 on facial and other physical morphology in miceY. Qian, Z. Xiong, Y. Li, and 3 more authorsFASEB Bioadv, 2021
DNA variants in or close to the human TBX15 and PAX1 genes have been repeatedly associated with facial morphology in independent genome-wide association studies, while their functional roles in determining facial morphology remain to be understood. We generated Tbx15 knockout (Tbx15 (-/-)) and Pax1 knockout (Pax1 (-/-)) mice by applying the one-step CRISPR/Cas9 method. A total of 75 adult mice were used for subsequent phenotype analysis, including 38 Tbx15 mice (10 homozygous Tbx15 (-/-), 18 heterozygous Tbx15 (+/-), 10 wild-type Tbx15 (+/+) WT littermates) and 37 Pax1 mice (12 homozygous Pax1 (-/-), 15 heterozygous Pax1 (+/-), 10 Pax1 (+/+) WT littermates). Facial and other physical morphological phenotypes were obtained from three-dimensional (3D) images acquired with the HandySCAN BLACK scanner. Compared to WT littermates, the Tbx15 (-/-) mutant mice had significantly shorter faces (p = 1.08E-8, R(2) = 0.61) and their ears were in a significantly lower position (p = 3.54E-8, R(2) = 0.62) manifesting a "droopy ear" characteristic. Besides these face alternations, Tbx15 (-/-) mutant mice displayed significantly lower weight as well as shorter body and limb length. Pax1 (-/-) mutant mice showed significantly longer noses (p = 1.14E-5, R(2) = 0.46) relative to WT littermates, but otherwise displayed less obvious morphological alterations than Tbx15 (-/-) mutant mice did. We provide the first direct functional evidence that two well-known and replicated human face genes, Tbx15 and Pax1, impact facial and other body morphology in mice. The general agreement between our findings in knock-out mice with those from previous GWASs suggests that the functional evidence we established here in mice may also be relevant in humans.
- SciAdvGenome-wide association study in almost 195,000 individuals identifies 50 previously unidentified genetic loci for eye colorM. Simcoe, A. Valdes, F. Liu, and 43 more authorsSci Adv, 2021
Human eye color is highly heritable, but its genetic architecture is not yet fully understood. We report the results of the largest genome-wide association study for eye color to date, involving up to 192,986 European participants from 10 populations. We identify 124 independent associations arising from 61 discrete genomic regions, including 50 previously unidentified. We find evidence for genes involved in melanin pigmentation, but we also find associations with genes involved in iris morphology and structure. Further analyses in 1636 Asian participants from two populations suggest that iris pigmentation variation in Asians is genetically similar to Europeans, albeit with smaller effect sizes. Our findings collectively explain 53.2% (95% confidence interval, 45.4 to 61.0%) of eye color variation using common single-nucleotide polymorphisms. Overall, our study outcomes demonstrate that the genetic complexity of human eye color considerably exceeds previous knowledge and expectations, highlighting eye color as a genetically highly complex human trait.
- IJLMDNA-based eyelid trait prediction in Chinese Han populationQ. Wang, B. Jin, F. Liu, and 4 more authorsInt J Legal Med, 2021
The eyelid folding represents one of the most distinguishing features of East Asian faces, involving the absence or presence of the eyelid crease, i.e., single vs. double eyelid. Recently, a genome-wide association study (GWAS) identified two SNPs (rs12570134 and rs1415425) showing genome-wide significant association with the double eyelid phenotype in Japanese. Here we report a confirmatory study in 697 Chinese individuals of exclusively Han origin. Only rs1415425 was statistically significant (P-value = 0.011), and the allele effect was on the same direction with that reported in Japanese. This SNP combined with gender and age explained 10.0% of the total variation in eyelid folding. DNA-based prediction model for the eyelid trait was developed and evaluated using logistic regression. The model showed mild to moderate predictive capacity (AUC = 0.69, sensitivity = 63%, and specificity = 70%). We further selected six additional SNPs by massive parallel sequencing of 19 candidate genes in 24 samples, and one SNP rs2761882 was statistically significant (P-value = 0.027). All predictors including these two SNPs (rs1415425 and rs2761882), gender, and age explained 11.2% of the total variation. The combined prediction model obtained an improved predictive capacity (AUC = 0.72, sensitivity = 62%, and specificity = 66%). Our study thus provided a confirmation of previous GWAS findings and a DNA-based prediction of the eyelid trait in Chinese Han individuals. This model may add value to forensic DNA phenotyping applications considering gender and age can be separately inferred from genetic and epigenetic markers. To further improve the prediction accuracy, future studies should focus on identifying more informative SNPs by large GWASs in East Asian populations.
2020
- FSIGExplaining sudden infant death with cardiac arrhythmias: Complete exon sequencing of nine cardiac arrhythmia genes in Dutch SIDS cases highlights new and known DNA variantsG. Liebrechts-Akkerman, F. Liu, R. Marion, and 2 more authorsForensic Sci Int Genet, 2020
Previous studies suggested that Sudden Infant Death Syndrome (SIDS) can partially be genetically explained by cardiac arrhythmias; however, the number of individuals and populations investigated remain limited. We report the first SIDS study on cardiac arrhythmias genes from the Netherlands, a country with the lowest SIDS incidence likely due to parent education on awareness of environmental risk factors. By using targeted massively parallel sequencing (MPS) in 142 Dutch SIDS cases, we performed a complete exon screening of all 173 exons from 9 cardiac arrhythmias genes SCN5A, KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1C, CAV3, ANK2 and KCNJ2 ( approximately 34,000 base pairs), that were selected to harbour previously established SIDS-associated DNA variants. Motivated by the poor DNA quality from the paraffin embedded material used, the application of a conservative sequencing quality control protocol resulted in 102 SIDS cases surviving quality control. Amongst the 102 SIDS cases, we identified a total of 40 DNA variants in 8 cardiac arrhythmia genes found in 60 (58.8 %) cases. Statistical analyses using ancestry-adjusted reference population data and multiple test correction revealed that 13 (32.5 %) of the identified DNA variants in 6 cardiac arrhythmia genes were significantly associated with SIDS, which were observed in 15 (14.7 %) SIDS cases. These 13, and another three, DNA variants were classified as likely pathogenic for cardiac arrhythmias using the American College of Medical Genetics guidelines for interpretation of sequence variants. The 16 likely pathogenic DNA variants were found in 16 (15.7 %) SIDS cases, including i) 3 novel DNA variants not recorded in public databases ii) 7 known DNA variants for which significant SIDS association established here was previously unknown, and iii) 6 known DNA variants for which LQTS association was reported previously. By having replicated previously reported SIDS-associated DNA variants located in cardiac arrhythmia genes and by having highlighting novel SIDS-associated DNA variants in such genes, our findings provide additional empirical evidence for the partial genetic explanation of SIDS by cardiac arrhythmias. On a wider abbr, our study outcome stresses the need for routine post-mortem genetic screening of assumed SIDS cases, particularly for cardiac arrhythmia genes. When put in practise, it will allow preventing further sudden deaths (not only in infants) in the affected families, thereby allowing forensic molecular autopsy not only to provide answers on the cause of death, but moreover to save lives.
- YiChuan[Human facial shape related SNP analysis in Han Chinese populations]M. Liu, Y. Li, Y. F. Yang, and 5 more authorsYi Chuan, 2020
Human facial morphology is one of the important visible biological characteristics. Understanding the genetic basis underlying facial shape traits has important implications in population genetics, developmental biology, and forensic science. This study extracted 136 Euclidean distance phenotypes from 17 facial features of high-resolution 3D facial images in 1177 Chinese Han adult males. Based on 3× low-depth sequencing data, linear regression was used to analyze the correlation between 125 reported SNPs significantly associated with facial morphology and 136 facial phenotypes. As a result, a total of twelve SNPs from ten genes demonstrated significant association with one or more facial shape traits after adjusting for multiple testing (significance threshold
- BrJDermatolPrincipal component analysis of seven skin-ageing features identifies three main types of skin ageingL. M. Pardo, M. A. Hamer, F. Liu, and 4 more authorsBr J Dermatol, 2020
BACKGROUND: The underlying phenotypic correlations between wrinkles, pigmented spots (PS), telangiectasia and other related facial-ageing subphenotypes are not well understood. OBJECTIVES: To analyse the underlying phenotypic correlation structure between seven features for facial ageing: global wrinkling, perceived age (PA), Griffiths photodamage grading, PS, telangiectasia, actinic keratosis (AK) and keratinocyte cancer (KC). METHODS: This was a cross-sectional study. Facial photographs and a full-body skin examination were used. We used principal component analysis (PCA) to derive principal components (PCs) of common variation between the features. We performed multivariable linear regressions between age, sex, body mass index, smoking and ultraviolet radiation exposure and the PC scores derived from PCA. We also tested the association between the main PC scores and 140 single-nucleotide polymorphisms (SNPs) previously associated with skin-ageing phenotypes. RESULTS: We analysed data from 1790 individuals with complete data on seven features of skin ageing. Three main PCs explained 73% of the total variance of the ageing phenotypes: a hypertrophic/wrinkling component (PC1: global wrinkling, PA and Griffiths grading), an atrophic/skin colour component (PC2: PS and telangiectasia) and a cancerous component (PC3: AK and KC). The associations between lifestyle and host factors differed per PC. The strength of SNP associations also differed per component with the most SNP associations found with the atrophic component [e.g. the IRF4 SNP (rs12203592); P-value = 1.84 x 10(-22) ]. CONCLUSIONS: Using a hypothesis-free approach, we identified three major underlying phenotypes associated with extrinsic ageing. Associations between determinants for skin ageing differed in magnitude and direction per component. What’s already known about this topic? Facial ageing is a complex phenotype consisting of different features including wrinkles, pigmented changes, telangiectasia and cancerous-related growths; it is not clear how these phenotypes are related to each other and to other phenotypes. A few studies have described two main clinical phenotypes for photoageing, namely hypertrophic ageing and atrophic ageing, which have been based solely on the clinical assessment of photoageing characteristics. What does this study add? We are the first to use epidemiology data to identify three main components associated with photoageing, namely a hypertrophic component (global wrinkling; perceived age; Griffiths grading) and atrophic component (pigmented spots; telangiectasia) and a cancer component (actinic keratosis; keratinocyte cancer). Association analysis showed different effects and direction of environmental determinants and genetic associations with the three components, with the most significant gene variants associations found for the atrophic component.
- ClinGenetA genome-wide association study identifies FSHR rs2300441 associated with follicle-stimulating hormone levelsJ. Yan, Y. Tian, X. Gao, and 8 more authorsClin Genet, 2020
Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) play critical roles in female reproduction, while the underlying genetic basis is poorly understood. Genome-wide association studies (GWASs) of FSH and LH levels were conducted in 2590 Chinese females including 1882 polycystic ovary syndrome (PCOS) cases and 708 controls. GWAS for FSH level identified multiple variants at FSHR showing genome-wide significance with the top variant (rs2300441) located in the intron of FSHR. The A allele of rs2300441 led to a reduced level of FSH in the PCOS group (beta = -.43, P = 6.70 x 10(-14) ) as well as in the control group (beta = -.35, P = 6.52 x 10(-4) ). In the combined sample, this association was enhanced after adjusting for the PCOS status (before: beta = -.38, P = 1.77 x 10(-13) ; after: beta = -.42, P = 3.33 x 10(-16) ), suggesting the genetic effect is independent of the PCOS status. The rs2300441 explained sevenfold higher proportion of the FSH variance than the total variance explained by the two previously reported FSHR missense variants (rs2300441 R(2) = 1.40% vs rs6166 R(2) = 0.17%, rs6165 R(2) = 0.03%). GWAS for LH did not identify any genome-wide significant associations. In conclusion, we identified genome-wide significant association between variants in FSHR and circulating FSH first, with the top associated variant rs2300441 might be a primary contributor at the population level.
2019
- GPBWhole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han GenomeZ. Du, L. Ma, H. Qu, and 27 more authorsGenomics Proteomics Bioinformatics, 2019
To unravel the genetic mechanisms of disease and physiological traits, it requires comprehensive sequencing analysis of large sample size in Chinese populations. Here, we report the primary results of the Chinese Academy of Sciences Precision Medicine Initiative (CASPMI) project launched by the Chinese Academy of Sciences, including the de novo assembly of a northern Han reference genome (NH1.0) and whole genome analyses of 597 healthy people coming from most areas in China. Given the two existing reference genomes for Han Chinese (YH and HX1) were both from the south, we constructed NH1.0, a new reference genome from a northern individual, by combining the sequencing strategies of PacBio, 10x Genomics, and Bionano mapping. Using this integrated approach, we obtained an N50 scaffold size of 46.63 Mb for the NH1.0 genome and performed a comparative genome analysis of NH1.0 with YH and HX1. In order to generate a genomic variation map of Chinese populations, we performed the whole-genome sequencing of 597 participants and identified 24.85 million (M) single nucleotide variants (SNVs), 3.85 M small indels, and 106,382 structural variations. In the association analysis with collected phenotypes, we found that the T allele of rs1549293 in KAT8 significantly correlated with the waist circumference in northern Han males. Moreover, significant genetic diversity in MTHFR, TCN2, FADS1, and FADS2, which associate with circulating folate, vitamin B12, or lipid metabolism, was observed between northerners and southerners. Especially, for the homocysteine-increasing allele of rs1801133 (MTHFR 677T), we hypothesize that there exists a "comfort" zone for a high frequency of 677T between latitudes of 35-45 degree North. Taken together, our results provide a high-quality northern Han reference genome and novel population-specific data sets of genetic variants for use in the personalized and precision medicine.
- IJLMPredicting adult height from DNA variants in a European-Asian admixed populationX. Jing, Y. Sun, W. Zhao, and 4 more authorsInt J Legal Med, 2019
Accurate genomic profiling for adult height is of high practical relevance in forensics genetics. Adult height is a classical reference trait in the field of human complex trait genetics characterized by highly polygenic nature and relatively high heritability. A meta-analysis of genome-wide association studies by the Genetic Investigation of Anthropocentric Traits (GIANT) consortium has identified 697 DNA variants associated with adult height in Europeans; however, whether these variants will still be informative in non-Europeans is still in question. The present study investigated the predictive power of these 697 height-associated SNPs in 687 Uyghurs of European-Asian admixed origin. Among all GIANT SNPs, 11% showed nominally significant association (6.78 x 10(-4) < p < 0.05) with adult height in the Uyghur population and among the significant SNPs 77% of allele effects were in the same direction as those in Europeans reported in the GIANT study. Fitting linear and logistic models using a polygenic score consisting of all GIANT SNPs resulted in an 80-20 cross-validated mean R(2) of 10.08% (95% CI 3.16-18.40%) for quantitative height prediction and a mean AUC value of 0.65 (95% CI 0.57-0.72%) for qualitative "above average" prediction. Fine-tuning the SNP set using their association p values considerably improved the prediction results (number of SNPs = 62, R(2) = 15.59%, 95% CI 6.80-25.71%; AUC = 0.70, 95% CI 62-0.77) in the Uyghurs. Overall, our findings demonstrate substantial differences between the European and Asian populations in the genetics of adult height, emphasizing the importance of population heterogeneity underlying the genetic architecture of adult height.
- HumGenetEDAR, LYPLAL1, PRDM16, PAX3, DKK1, TNFSF12, CACNA2D3, and SUPT3H gene variants influence facial morphology in a Eurasian populationY. Li, W. Zhao, D. Li, and 8 more authorsHum Genet, 2019
In human society, the facial surface is visible and recognizable based on the facial shape variation which represents a set of highly polygenic and correlated complex traits. Understanding the genetic basis underlying facial shape traits has important implications in population genetics, developmental biology, and forensic science. A number of single nucleotide polymorphisms (SNPs) are associated with human facial shape variation, mostly in European populations. To bridge the gap between European and Asian populations in term of the genetic basis of facial shape variation, we examined the effect of these SNPs in a European-Asian admixed Eurasian population which included a total of 612 individuals. The coordinates of 17 facial landmarks were derived from high resolution 3dMD facial images, and 136 Euclidean distances between all pairs of landmarks were quantitatively derived. DNA samples were genotyped using the Illumina Infinium Global Screening Array and imputed using the 1000 Genomes reference panel. Genetic association between 125 previously reported facial shape-associated SNPs and 136 facial shape phenotypes was tested using linear regression. As a result, a total of eight SNPs from different loci demonstrated significant association with one or more facial shape traits after adjusting for multiple testing (significance threshold p < 1.28 x 10(-3)), together explaining up to 6.47% of sex-, age-, and BMI-adjusted facial phenotype variance. These included EDAR rs3827760, LYPLAL1 rs5781117, PRDM16 rs4648379, PAX3 rs7559271, DKK1 rs1194708, TNFSF12 rs80067372, CACNA2D3 rs56063440, and SUPT3H rs227833. Notably, the EDAR rs3827760 and LYPLAL1 rs5781117 SNPs displayed significant association with eight and seven facial phenotypes, respectively (2.39 x 10(-5) < p < 1.28 x 10(-3)). The majority of these SNPs showed a distinct allele frequency between European and East Asian reference panels from the 1000 Genomes Project. These results showed the details of above eight genes influence facial shape variation in a Eurasian population.
- FYXZZPigmentation Phenotype Prediction of Chinese Populations from Different Language FamiliesQ. S. Liang, M. Liu, X. M. Tao, and 4 more authorsFa Yi Xue Za Zhi, 2019
Objective To predict the pigmentation phenotypes of Chinese populations from different language families, analyze the differences and provide reference data for forensic anthropology and genetics. Methods The HIrisPlex-S multiplex amplification system with 41 loci related to pigmentation phenotypes was constructed in the laboratory, and 2 666 DNA samples of adult males of 17 populations from six language families, including Indo-European, Sino-Tibetan, Altaic, Hmong-Mien, Tai-Kadai and Austro-Asiatic language families distributed in different regions of China were genotyped. The pigmentation phenotype category of each individual was predicted using the online prediction system (https://HIrisPlex.erasmusmc.nl/), and then the output data were statistically analyzed. Results About 1.92% of the individuals of Asian-European admixed populations from Indo-European and Altaic language families had blue eyes and 34.29% had brown or gold hair. The phenotypes of the color of eyes and hair of other populations had no significant difference, all individuals had brown eyes and black hair. There were differences in skin color of populations of different language families and geographical areas. The Indo-European language family had the lightest skin color, and the Austro-Asiatic language family had the darkest skin color; the southwestern minority populations had a darker skin color than populations in the plain areas. Conclusion The prediction results of pigmentation phenotype of Chinese populations are consistent with the perception of the appearance of each population, proving the reliability of the system. The color of eyes and hair are mainly related to ancestral components, while the skin color shows the differences between language families, and is closely related to geographical distribution of populations.
- FSIGUpdate on the predictability of tall stature from DNA markers in EuropeansF. Liu, K. Zhong, X. Jing, and 4 more authorsForensic Sci Int Genet, 2019
Predicting adult height from DNA has important implications in forensic DNA phenotyping. In 2014, we introduced a prediction model consisting of 180 height-associated SNPs based on data from 10,361 Northwestern Europeans enriched with tall individuals (770 > 1.88 standard deviation), which yielded a mid-ranged accuracy (AUC = 0.75 for binary prediction of tall stature and R(2) = 0.12 for quantitative prediction of adult height). Here, we provide an update on DNA-based height predictability considering an enlarged list of subsequently-published height-associated SNPs using data from the same set of 10,361 Europeans. A prediction model based on the full set of 689 SNPs showed an improved accuracy relative to previous models for both tall stature (AUC = 0.79) and quantitative height (R(2) = 0.21). A feature selection analysis revealed a subset of 412 most informative SNPs while the corresponding prediction model retained most of the accuracy (AUC = 0.76 and R(2) = 0.19) achieved with the full model. Over all, our study empirically exemplifies that the accuracy for predicting human appearance phenotypes with very complex underlying genetic architectures, such as adult height, can be improved by increasing the number of phenotype-associated DNA variants. Our work also demonstrates that a careful sub-selection allows for a considerable reduction of the number of DNA predictors that achieve similar prediction accuracy as provided by the full set. This is forensically relevant due to restrictions in the number of SNPs simultaneously analyzable with forensically suitable DNA technologies in the current days of targeted massively parallel sequencing in forensic genetics.
- EurJEpidemioValidated inference of smoking habits from blood with a finite DNA methylation marker setS. C. E. Maas, A. Vidaki, R. Wilson, and 26 more authorsEur J Epidemiol, 2019
Inferring a person’s smoking habit and history from blood is relevant for complementing or replacing self-reports in epidemiological and public health research, and for forensic applications. However, a finite DNA methylation marker set and a validated statistical model based on a large dataset are not yet available. Employing 14 epigenome-wide association studies for marker discovery, and using data from six population-based cohorts (N = 3764) for model building, we identified 13 CpGs most suitable for inferring smoking versus non-smoking status from blood with a cumulative Area Under the Curve (AUC) of 0.901. Internal fivefold cross-validation yielded an average AUC of 0.897 +/- 0.137, while external model validation in an independent population-based cohort (N = 1608) achieved an AUC of 0.911. These 13 CpGs also provided accurate inference of current (average AUC(crossvalidation) 0.925 +/- 0.021, AUC(externalvalidation)0.914), former (0.766 +/- 0.023, 0.699) and never smoking (0.830 +/- 0.019, 0.781) status, allowed inferring pack-years in current smokers (10 pack-years 0.800 +/- 0.068, 0.796; 15 pack-years 0.767 +/- 0.102, 0.752) and inferring smoking cessation time in former smokers (5 years 0.774 +/- 0.024, 0.760; 10 years 0.766 +/- 0.033, 0.764; 15 years 0.767 +/- 0.020, 0.754). Model application to children revealed highly accurate inference of the true non-smoking status (6 years of age: accuracy 0.994, N = 355; 10 years: 0.994, N = 309), suggesting prenatal and passive smoking exposure having no impact on model applications in adults. The finite set of DNA methylation markers allow accurate inference of smoking habit, with comparable accuracy as plasma cotinine use, and smoking history from blood, which we envision becoming useful in epidemiology and public health research, and in medical and forensic applications.
- FSIGValidation of methylation-based forensic age estimation in time-series bloodstains on FTA cards and gauze at room temperature conditionsF. Peng, L. Feng, J. Chen, and 6 more authorsForensic Sci Int Genet, 2019
We previously proposed a prediction model consisting of 9 CpG sites for forensic age estimation with high practical potentials in Chinese males. Here, we further evaluated the performance of this prediction model in two independent batches of time-series bloodstain samples naturally exposed to room temperature conditions. The first batch consists of 30 Han Chinese males (18-59 years of age) whose peripheral blood was converted into bloodstains on Flinders Technology Association (FTA) cards and naturally exposed to room temperature conditions for different time points up to 3 months. The second batch consists of 99 Han Chinese males (21-66 years of age) whose peripheral blood was divided into 3 replicates, converted into bloodstains on gauze, and naturally exposed to room temperature conditions for 3 months. For each time point and each replicate, the methylation levels at the 9 CpG sites were detected using the EpiTYPER system. Applying the 9-CpG age prediction model to these bloodstain samples resulted in highly accurate age predictions for all time points and replicates (0.81 <R(2) < 0.91, 2.94 < MAD < 3.55 years). The updated model combining our previous and current data achieved similarly high prediction results. Therefore, our 9-CpG age prediction model was successfully validated in time-series bloodstain samples converted on both FTA card and gauze under natural room temperature conditions, demonstrating high potentials in future forensic applications to Han Chinese males.
- ElifeNovel genetic loci affecting facial shape variation in humansZ. Xiong, G. Dankova, L. J. Howe, and 47 more authorsElife, 2019
The human face represents a combined set of highly heritable phenotypes, but knowledge on its genetic architecture remains limited, despite the relevance for various fields. A series of genome-wide association studies on 78 facial shape phenotypes quantified from 3-dimensional facial images of 10,115 Europeans identified 24 genetic loci reaching study-wide suggestive association (p < 5 x 10(-8)), among which 17 were previously unreported. A follow-up multi-ethnic study in additional 7917 individuals confirmed 10 loci including six unreported ones (p(adjusted) < 2.1 x 10(-3)). A global map of derived polygenic face scores assembled facial features in major continental groups consistent with anthropological knowledge. Analyses of epigenomic datasets from cranial neural crest cells revealed abundant cis-regulatory activities at the face-associated genetic loci. Luciferase reporter assays in neural crest progenitor cells highlighted enhancer activities of several face-associated DNA variants. These results substantially advance our understanding of the genetic basis underlying human facial variation and provide candidates for future in-vivo functional studies.
2018
- FSIGThe HIrisPlex-S system for eye, hair and skin colour prediction from DNA: Introduction and forensic developmental validationL. Chaitanya, K. Breslin, S. Zuniga, and 9 more authorsForensic Sci Int Genet, 2018
Forensic DNA Phenotyping (FDP), i.e. the prediction of human externally visible traits from DNA, has become a fast growing subfield within forensic genetics due to the intelligence information it can provide from DNA traces. FDP outcomes can help focus police investigations in search of unknown perpetrators, who are generally unidentifiable with standard DNA profiling. Therefore, we previously developed and forensically validated the IrisPlex DNA test system for eye colour prediction and the HIrisPlex system for combined eye and hair colour prediction from DNA traces. Here we introduce and forensically validate the HIrisPlex-S DNA test system (S for skin) for the simultaneous prediction of eye, hair, and skin colour from trace DNA. This FDP system consists of two SNaPshot-based multiplex assays targeting a total of 41 SNPs via a novel multiplex assay for 17 skin colour predictive SNPs and the previous HIrisPlex assay for 24 eye and hair colour predictive SNPs, 19 of which also contribute to skin colour prediction. The HIrisPlex-S system further comprises three statistical prediction models, the previously developed IrisPlex model for eye colour prediction based on 6 SNPs, the previous HIrisPlex model for hair colour prediction based on 22 SNPs, and the recently introduced HIrisPlex-S model for skin colour prediction based on 36 SNPs. In the forensic developmental validation testing, the novel 17-plex assay performed in full agreement with the Scientific Working Group on DNA Analysis Methods (SWGDAM) guidelines, as previously shown for the 24-plex assay. Sensitivity testing of the 17-plex assay revealed complete SNP profiles from as little as 63 pg of input DNA, equalling the previously demonstrated sensitivity threshold of the 24-plex HIrisPlex assay. Testing of simulated forensic casework samples such as blood, semen, saliva stains, of inhibited DNA samples, of low quantity touch (trace) DNA samples, and of artificially degraded DNA samples as well as concordance testing, demonstrated the robustness, efficiency, and forensic suitability of the new 17-plex assay, as previously shown for the 24-plex assay. Finally, we provide an update to the publically available HIrisPlex website https://hirisplex.erasmusmc.nl/, now allowing the estimation of individual probabilities for 3 eye, 4 hair, and 5 skin colour categories from HIrisPlex-S input genotypes. The HIrisPlex-S DNA test represents the first forensically validated tool for skin colour prediction, and reflects the first forensically validated tool for simultaneous eye, hair and skin colour prediction from DNA.
- NatCommunNovel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathwaysD. L. Duffy, G. Zhu, X. Li, and 42 more authorsNat Commun, 2018
The total number of acquired melanocytic nevi on the skin is strongly correlated with melanoma risk. Here we report a meta-analysis of 11 nevus GWAS from Australia, Netherlands, UK, and USA comprising 52,506 individuals. We confirm known loci including MTAP, PLA2G6, and IRF4, and detect novel SNPs in KITLG and a region of 9q32. In a bivariate analysis combining the nevus results with a recent melanoma GWAS meta-analysis (12,874 cases, 23,203 controls), SNPs near GPRC5A, CYP1B1, PPARGC1B, HDAC4, FAM208B, DOCK8, and SYNE2 reached global significance, and other loci, including MIR146A and OBFC1, reached a suggestive level. Overall, we conclude that most nevus genes affect melanoma risk (KITLG an exception), while many melanoma risk loci do not alter nevus count. For example, variants in TERC and OBFC1 affect both traits, but other telomere length maintenance genes seem to affect melanoma risk only. Our findings implicate multiple pathways in nevogenesis.
- FSIGSystematic feature selection improves accuracy of methylation-based forensic age estimation in Han Chinese malesL. Feng, F. Peng, S. Li, and 6 more authorsForensic Sci Int Genet, 2018
Estimating individual age from biomarkers may provide key information facilitating forensic investigations. Recent progress has shown DNA methylation at age-associated CpG sites as the most informative biomarkers for estimating the individual age of an unknown donor. Optimal feature selection plays a critical role in determining the performance of the final prediction model. In this study we investigate methylation levels at 153 age-associated CpG sites from 21 previously reported genomic regions using the EpiTYPER system for their predictive power on individual age in 390 Han Chinese males ranging from 15 to 75 years of age. We conducted a systematic feature selection using a stepwise backward multiple linear regression analysis as well as an exhaustive searching algorithm. Both approaches identified the same subset of 9 CpG sites, which in linear combination provided the optimal model fitting with mean absolute deviation (MAD) of 2.89 years of age and explainable variance (R(2)) of 0.92. The final model was validated in two independent Han Chinese male samples (validation set 1, N = 65, MAD = 2.49, R(2) = 0.95, and validation set 2, N = 62, MAD = 3.36, R(2) = 0.89). Other competing models such as support vector machine and artificial neural network did not outperform the linear model to any noticeable degree. The validation set 1 was additionally analyzed using Pyrosequencing technology for cross-platform validation and was termed as validation set 3. Directly applying our model, in which the methylation levels were detected by the EpiTYPER system, to the data from pyrosequencing technology showed, however, less accurate results in terms of MAD (validation set 3, N = 65 Han Chinese males, MAD = 4.20, R(2) = 0.93), suggesting the presence of a batch effect between different data generation platforms. This batch effect could be partially overcome by a z-score transformation (MAD = 2.76, R(2) = 0.93). Overall, our systematic feature selection identified 9 CpG sites as the optimal subset for forensic age estimation and the prediction model consisting of these 9 markers demonstrated high potential in forensic practice. An age estimator implementing our prediction model allowing missing markers is freely available at http://liufan.big.ac.cn/AgePrediction.
- NatGenet
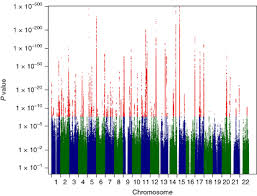 Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritabilityP. G. Hysi, A. M. Valdes, F. Liu, and 42 more authorsNat Genet, 2018
Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritabilityP. G. Hysi, A. M. Valdes, F. Liu, and 42 more authorsNat Genet, 2018Hair color is one of the most recognizable visual traits in European populations and is under strong genetic control. Here we report the results of a genome-wide association study meta-analysis of almost 300,000 participants of European descent. We identified 123 autosomal and one X-chromosome loci significantly associated with hair color; all but 13 are novel. Collectively, single-nucleotide polymorphisms associated with hair color within these loci explain 34.6% of red hair, 24.8% of blond hair, and 26.1% of black hair heritability in the study populations. These results confirm the polygenic nature of complex phenotypes and improve our understanding of melanin pigment metabolism in humans.
- FYXZZ[Assessment of Height Prediction Model Based on SNPs Loci]H. Y. Jiao, Y. N. Sun, X. X. Jing, and 7 more authorsFa Yi Xue Za Zhi, 2018
OBJECTIVES: To establish a height prediction model of Chinese Han male based on the reported 547 height-associated single nucleotide polymorphisms (SNPs) loci in Europeans, and assess its accuracy for height estimation. METHODS: The DNA typing was analyzed in 59 Han male samples of Shandong province by Affymetrix SNP Array 6.0 chip and HiSeq 4000 sequencing platform. Prediction model was established using 547 height-associated SNPs loci as predictors and weight allele sums (WAS) as computing method. The accuracy of height prediction model was analysed using receiver operating characteristic (ROC) curve and area under curve (AUC). RESULTS: There was no height-associated SNPs locus was found by genome-wide association studies. In present study, height prediction model was established by WAS and obtained an AUC of 0.67 (95% CI: 0.53-0.90). CONCLUSIONS: It has reference value for predicting the height of Han male in Shandong province by WAS model based on 547 SNPs loci, while it is still necessary to further promote the accuracy of the prediction model by screening more height-associated SNPs loci with population heterogeneity.
- IJLMInvestigation of metabolites for estimating blood deposition timeK. Lech, F. Liu, S. K. Davies, and 9 more authorsInt J Legal Med, 2018
Trace deposition timing reflects a novel concept in forensic molecular biology involving the use of rhythmic biomarkers for estimating the time within a 24-h day/night cycle a human biological sample was left at the crime scene, which in principle allows verifying a sample donor’s alibi. Previously, we introduced two circadian hormones for trace deposition timing and recently demonstrated that messenger RNA (mRNA) biomarkers significantly improve time prediction accuracy. Here, we investigate the suitability of metabolites measured using a targeted metabolomics approach, for trace deposition timing. Analysis of 171 plasma metabolites collected around the clock at 2-h intervals for 36 h from 12 male participants under controlled laboratory conditions identified 56 metabolites showing statistically significant oscillations, with peak times falling into three day/night time categories: morning/noon, afternoon/evening and night/early morning. Time prediction modelling identified 10 independently contributing metabolite biomarkers, which together achieved prediction accuracies expressed as AUC of 0.81, 0.86 and 0.90 for these three time categories respectively. Combining metabolites with previously established hormone and mRNA biomarkers in time prediction modelling resulted in an improved prediction accuracy reaching AUCs of 0.85, 0.89 and 0.96 respectively. The additional impact of metabolite biomarkers, however, was rather minor as the previously established model with melatonin, cortisol and three mRNA biomarkers achieved AUC values of 0.88, 0.88 and 0.95 for the same three time categories respectively. Nevertheless, the selected metabolites could become practically useful in scenarios where RNA marker information is unavailable such as due to RNA degradation. This is the first metabolomics study investigating circulating metabolites for trace deposition timing, and more work is needed to fully establish their usefulness for this forensic purpose.
- YiChuan[The effect of EDARV370A on facial and ear morphologies in Uyghur population]Y. Li, W. T. Zhao, D. Li, and 9 more authorsYi Chuan, 2018
The ectodysplasinA receptor gene (EDAR) plays an important role in the development of ectoderm. The derived G allele of its key missense variant EDARV370A is prevalent in East Asians and Americans, but rare in Africans and Europeans. This leads to distinct ectodermal-derived phenotypes between different continental groups, such as the straighter and thicker hair, more eccrine sweat glands, feminine smaller breasts, shovel incisors characteristic of East Asians. At present, we know little about the association between EDARV370A and facial and ear morphology characteristics. To better understand the effect of EDARV370A on craniofacial phenotypes, we systematically examined the association between EDARV370A and 136 facial quantitative phenotypes, one chin ordinal phenotype and six ear ordinal phenotypes in 715 Uyghurs. The quantitative phenotypes were derived by applying our automated landmark annotation method to facial 3D photos and the ordinal phenotypes were manually graded from facial 2D photos. The analysis identified significant association (P<0.05 after multiple testing correction) between EDARV370A and eight facial phenotypes, one chin phenotype and three ear morphology phenotypes. Our study thus elucidated the pleotropic effect of EDARV370A on craniofacial phenotypes in a European-Asian admixed Uyghur population.
- HumMolGenetMeta-analysis of genome-wide association studies identifies 8 novel loci involved in shape variation of human head hairF. Liu, Y. Chen, G. Zhu, and 31 more authorsHum Mol Genet, 2018
Shape variation of human head hair shows striking variation within and between human populations, while its genetic basis is far from being understood. We performed a series of genome-wide association studies (GWASs) and replication studies in a total of 28 964 subjects from 9 cohorts from multiple geographic origins. A meta-analysis of three European GWASs identified 8 novel loci (1p36.23 ERRFI1/SLC45A1, 1p36.22 PEX14, 1p36.13 PADI3, 2p13.3 TGFA, 11p14.1 LGR4, 12q13.13 HOXC13, 17q21.2 KRTAP, and 20q13.33 PTK6), and confirmed 4 previously known ones (1q21.3 TCHH/TCHHL1/LCE3E, 2q35 WNT10A, 4q21.21 FRAS1, and 10p14 LINC00708/GATA3), all showing genome-wide significant association with hair shape (P < 5e-8). All except one (1p36.22 PEX14) were replicated with nominal significance in at least one of the 6 additional cohorts of European, Native American and East Asian origins. Three additional previously known genes (EDAR, OFCC1, and PRSS53) were confirmed at the nominal significance level. A multivariable regression model revealed that 14 SNPs from different genes significantly and independently contribute to hair shape variation, reaching a cross-validated AUC value of 0.66 (95% CI: 0.62-0.70) and an AUC value of 0.64 in an independent validation cohort, providing an improved accuracy compared with a previous model. Prediction outcomes of 2504 individuals from a multiethnic sample were largely consistent with general knowledge on the global distribution of hair shape variation. Our study thus delivers target genes and DNA variants for future functional studies to further evaluate the molecular basis of hair shape in humans.
- FSIGTowards broadening Forensic DNA Phenotyping beyond pigmentation: Improving the prediction of head hair shape from DNAE. Pospiech, Y. Chen, M. Kukla-Bartoszek, and 35 more authorsForensic Sci Int Genet, 2018
Human head hair shape, commonly classified as straight, wavy, cwebsitey or frizzy, is an attractive target for Forensic DNA Phenotyping and other applications of human appearance prediction from DNA such as in paleogenetics. The genetic knowledge underlying head hair shape variation was recently improved by the outcome of a series of genome-wide association and replication studies in a total of 26,964 subjects, highlighting 12 loci of which 8 were novel and introducing a prediction model for Europeans based on 14 SNPs. In the present study, we evaluated the capacity of DNA-based head hair shape prediction by investigating an extended set of candidate SNP predictors and by using an independent set of samples for model validation. Prediction model building was carried out in 9674 subjects (6068 from Europe, 2899 from Asia and 707 of admixed European and Asian ancestries), used previously, by considering a novel list of 90 candidate SNPs. For model validation, genotype and phenotype data were newly collected in 2415 independent subjects (2138 Europeans and 277 non-Europeans) by applying two targeted massively parallel sequencing platforms, Ion Torrent PGM and MiSeq, or the MassARRAY platform. A binomial model was developed to predict straight vs. non-straight hair based on 32 SNPs from 26 genetic loci we identified as significantly contributing to the model. This model achieved prediction accuracies, expressed as AUC, of 0.664 in Europeans and 0.789 in non-Europeans; the statistically significant difference was explained mostly by the effect of one EDAR SNP in non-Europeans. Considering sex and age, in addition to the SNPs, slightly and insignificantly increased the prediction accuracies (AUC of 0.680 and 0.800, respectively). Based on the sample size and candidate DNA markers investigated, this study provides the most robust, validated, and accurate statistical prediction models and SNP predictor marker sets currently available for predicting head hair shape from DNA, providing the next step towards broadening Forensic DNA Phenotyping beyond pigmentation traits.
- NatCommunGenome-wide association study in 176,678 Europeans reveals genetic loci for tanning response to sun exposureA. Visconti, D. L. Duffy, F. Liu, and 19 more authorsNat Commun, 2018
The skin’s tendency to sunburn rather than tan is a major risk factor for skin cancer. Here we report a large genome-wide association study of ease of skin tanning in 176,678 subjects of European ancestry. We identify significant association with tanning ability at 20 loci. We confirm previously identified associations at six of these loci, and report 14 novel loci, of which ten have never been associated with pigmentation-related phenotypes. Our results also suggest that variants at the AHR/AGR3 locus, previously associated with cutaneous malignant melanoma the underlying mechanism of which is poorly understood, might act on disease risk through modulation of tanning ability.
- PLoSGeneGenome-wide association studies and CRISPR/Cas9-mediated gene editing identify regulatory variants influencing eyebrow thickness in humansS. Wu, M. Zhang, X. Yang, and 33 more authorsPLoS Genet, 2018
Hair plays an important role in primates and is clearly subject to adaptive selection. While humans have lost most facial hair, eyebrows are a notable exception. Eyebrow thickness is heritable and widely believed to be subject to sexual selection. Nevertheless, few genomic studies have explored its genetic basis. Here, we performed a genome-wide scan for eyebrow thickness in 2961 Han Chinese. We identified two new loci of genome-wide significance, at 3q26.33 near SOX2 (rs1345417: P = 6.51x10(-10)) and at 5q13.2 near FOXD1 (rs12651896: P = 1.73x10(-8)). We further replicated our findings in the Uyghurs, a population from China characterized by East Asian-European admixture (N = 721), the CANDELA cohort from five Latin American countries (N = 2301), and the Rotterdam Study cohort of Dutch Europeans (N = 4411). A meta-analysis combining the full GWAS results from the three cohorts of full or partial Asian descent (Han Chinese, Uyghur and Latin Americans, N = 5983) highlighted a third signal of genome-wide significance at 2q12.3 (rs1866188: P = 5.81x10(-11)) near EDAR. We performed fine-mapping and prioritized four variants for further experimental verification. CRISPR/Cas9-mediated gene editing provided evidence that rs1345417 and rs12651896 affect the transcriptional activity of the nearby SOX2 and FOXD1 genes, which are both involved in hair development. Finally, suitable statistical analyses revealed that none of the associated variants showed clear signals of selection in any of the populations tested. Contrary to popular speculation, we found no evidence that eyebrow thickness is subject to strong selective pressure.

2017
- FSIGLikelihood ratio and posterior odds in forensic genetics: Two sides of the same coinA. Caliebe, S. Walsh, F. Liu, and 2 more authorsForensic Sci Int Genet, 2017
It has become widely accepted in forensics that, owing to a lack of sensible priors, the evidential value of matching DNA profiles in trace donor identification or kinship analysis is most sensibly communicated in the form of a likelihood ratio (LR). This restraint does not abate the fact that the posterior odds (PO) would be the preferred basis for returning a verdict. A completely different situation holds for Forensic DNA Phenotyping (FDP), which is aimed at predicting externally visible characteristics (EVCs) of a trace donor from DNA left behind at the crime scene. FDP is intended to provide leads to the police investigation helping them to find unknown trace donors that are unidentifiable by DNA profiling. The statistical models underlying FDP typically yield posterior odds (PO) for an individual possessing a certain EVC. This apparent discrepancy has led to confusion as to when LR or PO is the appropriate outcome of forensic DNA analysis to be communicated to the investigating authorities. We thus set out to clarify the distinction between LR and PO in the context of forensic DNA profiling and FDP from a statistical point of view. In so doing, we also addressed the influence of population affiliation on LR and PO. In contrast to the well-known population dependency of the LR in DNA profiling, the PO as obtained in FDP may be widely population-independent. The actual degree of independence, however, is a matter of (i) how much of the causality of the respective EVC is captured by the genetic markers used for FDP and (ii) by the extent to which non-genetic such as environmental causal factors of the same EVC are distributed equally throughout populations. The fact that an LR should be communicated in cases of DNA profiling whereas the PO are suitable for FDP does not conflict with theory, but rather reflects the immanent differences between these two forensic applications of DNA information.
- SciRepPredicting hair cortisol levels with hair pigmentation genes: a possible hair pigmentation biasA. Neumann, G. Noppe, F. Liu, and 5 more authorsSci Rep, 2017
Cortisol concentrations in hair are used to create hormone profiles spanning months. This method allows assessment of chronic cortisol exposure, but might be biased by hair pigmentation: dark hair was previously related to higher concentrations. It is unclear whether this association arises from local effects, such as increased hormone extractability, or whether the association represents systemic differences arising from population stratification. We tested the hypothesis that hair pigmentation gene variants are associated with varying cortisol levels independent of genetic ancestry. Hormone concentrations and genotype were measured in 1674 children from the Generation R cohort at age 6. We computed a polygenic score of hair color based on 9 single nucleotide polymorphisms. This score was used to predict hair cortisol concentrations, adjusted for genetic ancestry, sex, age and corticosteroid use. A 1-standard deviation (SD) higher polygenic score (darker hair) was associated with 0.08 SD higher cortisol levels (SE = 0.03, p = 0.002). This suggests that variation in hair cortisol concentrations is partly explained by local hair effects. In multi-ancestry studies this hair pigmentation bias can reduce power and confound results. Researchers should therefore consider adjusting analyses by reported hair color, by polygenic scores, or by both.
- HumGenetGlobal skin colour prediction from DNAS. Walsh, L. Chaitanya, K. Breslin, and 9 more authorsHum Genet, 2017
Human skin colour is highly heritable and externally visible with relevance in medical, forensic, and anthropological genetics. Although eye and hair colour can already be predicted with high accuracies from small sets of carefully selected DNA markers, knowledge about the genetic predictability of skin colour is limited. Here, we investigate the skin colour predictive value of 77 single-nucleotide polymorphisms (SNPs) from 37 genetic loci previously associated with human pigmentation using 2025 individuals from 31 global populations. We identified a minimal set of 36 highly informative skin colour predictive SNPs and developed a statistical prediction model capable of skin colour prediction on a global scale. Average cross-validated prediction accuracies expressed as area under the receiver-operating characteristic curve (AUC) +/- standard deviation were 0.97 +/- 0.02 for Light, 0.83 +/- 0.11 for Dark, and 0.96 +/- 0.03 for Dark-Black. When using a 5-category, this resulted in 0.74 +/- 0.05 for Very Pale, 0.72 +/- 0.03 for Pale, 0.73 +/- 0.03 for Intermediate, 0.87+/-0.1 for Dark, and 0.97 +/- 0.03 for Dark-Black. A comparative analysis in 194 independent samples from 17 populations demonstrated that our model outperformed a previously proposed 10-SNP-classifier approach with AUCs rising from 0.79 to 0.82 for White, comparable at the intermediate level of 0.63 and 0.62, respectively, and a large increase from 0.64 to 0.92 for Black. Overall, this study demonstrates that the chosen DNA markers and prediction model, particularly the 5-category level; allow skin colour predictions within and between continental regions for the first time, which will serve as a valuable resource for future applications in forensic and anthropologic genetics.
- SciRepNovel quantitative pigmentation phenotyping enhances genetic association, epistasis, and prediction of human eye colourA. Wollstein, S. Walsh, F. Liu, and 11 more authorsSci Rep, 2017
Success of genetic association and the prediction of phenotypic traits from DNA are known to depend on the accuracy of phenotype characterization, amongst other parameters. To overcome limitations in the characterization of human iris pigmentation, we introduce a fully automated approach that specifies the areal proportions proposed to represent differing pigmentation types, such as pheomelanin, eumelanin, and non-pigmented areas within the iris. We demonstrate the utility of this approach using high-resolution digital eye imagery and genotype data from 12 selected SNPs from over 3000 European samples of seven populations that are part of the EUREYE study. In comparison to previous quantification approaches, (1) we achieved an overall improvement in eye colour phenotyping, which provides a better separation of manually defined eye colour categories. (2) Single nucleotide polymorphisms (SNPs) known to be involved in human eye colour variation showed stronger associations with our approach. (3) We found new and confirmed previously abbrd SNP-SNP interactions. (4) We increased SNP-based prediction accuracy of quantitative eye colour. Our findings exemplify that precise quantification using the perceived biological basis of pigmentation leads to enhanced genetic association and prediction of eye colour. We expect our approach to deliver new pigmentation genes when applied to genome-wide association testing.
- HumGenetGenome-wide compound heterozygote analysis highlights alleles associated with adult height in EuropeansK. Zhong, G. Zhu, X. Jing, and 9 more authorsHum Genet, 2017
Adult height is the most widely genetically studied common trait in humans; however, the trait variance explainable by currently known height-associated single nucleotide polymorphisms (SNPs) identified from the previous genome-wide association studies (GWAS) is yet far from complete given the high heritability of this complex trait. To exam if compound heterozygotes (CH) may explain extra height variance, we conducted a genome-wide analysis to screen for CH in association with adult height in 10,631 Dutch Europeans enriched with extremely tall people, using our recently developed method implemented in the software package CollapsABEL. The analysis identified six regions (3q23, 5q35.1, 6p21.31, 6p21.33, 7q21.2, and 9p24.3), where multiple pairs of SNPs as CH showed genome-wide significant association with height (P < 1.67 x 10(-10)). Of those, 9p24.3 represents a novel region influencing adult height, whereas the others have been highlighted in the previous GWAS on height based on analysis of individual SNPs. A replication analysis in 4080 Australians of European ancestry confirmed the significant CH-like association at 9p24.3 (P < 0.05). Together, the collapsed genotypes at these six loci explained 2.51% of the height variance (after adjusting for sex and age), compared with 3.23% explained by the 14 top-associated SNPs at 14 loci identified by traditional GWAS in the same data set (P < 5 x 10(-8)). Overall, our study empirically demonstrates that CH plays an important role in adult height and may explain a proportion of its "missing heritability". Moreover, our findings raise promising expectations for other highly polygenic complex traits to explain missing heritability identifiable through CH-like associations.
2016
- FSIGEvaluation of mRNA markers for estimating blood deposition time: Towards alibi testing from human forensic stains with rhythmic biomarkersK. Lech, F. Liu, K. Ackermann, and 4 more authorsForensic Sci Int Genet, 2016
Determining the time a biological trace was left at a scene of crime reflects a crucial aspect of forensic investigations as - if possible - it would permit testing the sample donor’s alibi directly from the trace evidence, helping to link (or not) the DNA-identified sample donor with the crime event. However, reliable and robust methodology is lacking thus far. In this study, we assessed the suitability of mRNA for the purpose of estimating blood deposition time, and its added value relative to melatonin and cortisol, two circadian hormones we previously introduced for this purpose. By analysing 21 candidate mRNA markers in blood samples from 12 individuals collected around the clock at 2h intervals for 36h under real-life, controlled conditions, we identified 11 mRNAs with statistically significant expression rhythms. We then used these 11 significantly rhythmic mRNA markers, with and without melatonin and cortisol also analysed in these samples, to establish statistical models for predicting day/night time categories. We found that although in general mRNA-based estimation of time categories was less accurate than hormone-based estimation, the use of three mRNA markers HSPA1B, MKNK2 and PER3 together with melatonin and cortisol generally enhanced the time prediction accuracy relative to the use of the two hormones alone. Our data best support a model that by using these five molecular biomarkers estimates three time categories, i.e. night/early morning, morning/noon, and afternoon/evening with prediction accuracies expressed as AUC values of 0.88, 0.88, and 0.95, respectively. For the first time, we demonstrate the value of mRNA for blood deposition timing and introduce a statistical model for estimating day/night time categories based on molecular biomarkers, which shall be further validated with additional samples in the future. Moreover, our work provides new leads for molecular approaches on time of death estimation using the significantly rhythmic mRNA markers established here.
- CurrBiol
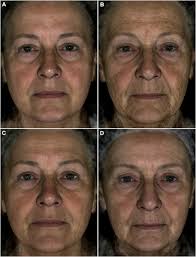 The MC1R Gene and Youthful LooksF. Liu, M. A. Hamer, J. Deelen, and 17 more authorsCurr Biol, 2016
The MC1R Gene and Youthful LooksF. Liu, M. A. Hamer, J. Deelen, and 17 more authorsCurr Biol, 2016Looking young for one’s age has been a desire since time immemorial. This desire is attributable to the belief that appearance reflects health and fecundity. Indeed, perceived age predicts survival [1] and associates with molecular markers of aging such as telomere length [2]. Understanding the underlying molecular biology of perceived age is vital for identifying new aging therapies among other purposes, but studies are lacking thus far. As a first attempt, we performed genome-wide association studies (GWASs) of perceived facial age and wrinkling estimated from digital facial images by analyzing over eight million SNPs in 2,693 elderly Dutch Europeans from the Rotterdam Study. The strongest genetic associations with perceived facial age were found for multiple SNPs in the MC1R gene (p < 1 x 10(-7)). This effect was enhanced for a compound heterozygosity marker constructed from four pre-selected functional MC1R SNPs (p = 2.69 x 10(-12)), which was replicated in 599 Dutch Europeans from the Leiden Longevity Study (p = 0.042) and in 1,173 Europeans of the TwinsUK Study (p = 3 x 10(-3)). Individuals carrying the homozygote MC1R risk haplotype looked on average up to 2 years older than non-carriers. This association was independent of age, sex, skin color, and sun damage (wrinkling, pigmented spots) and persisted through different sun-exposure levels. Hence, a role for MC1R in youthful looks independent of its known melanin synthesis function is suggested. Our study uncovers the first genetic evidence explaining why some people look older for their age and provides new leads for further investigating the biological basis of how old or young people look.
- EJHGPrediction of male-pattern baldness from genotypesF. Liu, M. A. Hamer, S. Heilmann, and 8 more authorsEur J Hum Genet, 2016
The global demand for products that effectively prevent the development of male-pattern baldness (MPB) has drastically increased. However, there is currently no established genetic model for the estimation of MPB risk. We conducted a prediction analysis using single-nucleotide polymorphisms (SNPs) identified from previous GWASs of MPB in a total of 2725 German and Dutch males. A logistic regression model considering the genotypes of 25 SNPs from 12 genomic loci demonstrates that early-onset MPB risk is predictable at an accuracy level of 0.74 when 14 SNPs were included in the model, and measured using the area under the receiver-operating characteristic curves (AUC). Considering age as an additional predictor, the model can predict normal MPB status in middle-aged and elderly individuals at a slightly lower accuracy (AUC 0.69-0.71) when 6-11 SNPs were used. A variance partitioning analysis suggests that 55.8% of early-onset MPB genetic liability can be explained by common autosomal SNPs and 23.3% by X-chromosome SNPs. For normal MPB status in elderly individuals, the proportion of explainable variance is lower (42.4% for autosomal and 9.8% for X-chromosome SNPs). The gap between GWAS findings and the variance partitioning results could be explained by a large body of common DNA variants with small effects that will likely be identified in GWAS of increased sample sizes. Although the accuracy obtained here has not reached a clinically desired level, our model was highly informative for up to 19% of Europeans, thus may assist decision making on early MPB intervention actions and in forensic investigations.
- BMC-BioinformaticsCollapsABEL: an R library for detecting compound heterozygote alleles in genome-wide association studiesK. Zhong, L. C. Karssen, M. Kayser, and 1 more authorBMC Bioinformatics, 2016
BACKGROUND: Compound Heterozygosity (CH) in classical genetics is the presence of two different recessive mutations at a particular gene locus. A relaxed form of CH alleles may account for an essential proportion of the missing heritability, i.e. heritability of phenotypes so far not accounted for by single genetic variants. Methods to detect CH-like effects in genome-wide association studies (GWAS) may facilitate explaining the missing heritability, but to our knowledge no viable software tools for this purpose are currently available. RESULTS: In this work we present the Generalized Compound Double Heterozygosity (GCDH) test and its implementation in the R package CollapsABEL. Time-consuming procedures are optimized for computational efficiency using Java or C++. Intermediate results are stored either in an SQL database or in a so-called big.matrix file to achieve reasonable memory footprint. Our large scale simulation studies show that GCDH is capable of discovering genetic associations due to CH-like interactions with much higher power than a conventional single-SNP approach under various settings, whether the causal genetic variations are available or not. CollapsABEL provides a user-friendly pipeline for genotype collapsing, statistical testing, power estimation, type I error control and graphics generation in the R language. CONCLUSIONS: CollapsABEL provides a computationally efficient solution for screening general forms of CH alleles in densely imputed microarray or whole genome sequencing datasets. The GCDH test provides an improved power over single-SNP based methods in detecting the prevalence of CH in human complex phenotypes, offering an opportunity for tackling the missing heritability problem. Binary and source packages of CollapsABEL are available on CRAN ( https://cran.r-project.org/web/packages/CollapsABEL ) and the website of the GenABEL project ( http://www.genabel.org/packages ).
- FSIGHuman age estimation from blood using mRNA, DNA methylation, DNA rearrangement, and telomere lengthD. Zubakov, F. Liu, I. Kokmeijer, and 9 more authorsForensic Sci Int Genet, 2016
Establishing the age of unknown persons, or persons with unknown age, can provide important leads in police investigations, disaster victim identification, fraud cases, and in other legal affairs. Previous methods mostly relied on morphological features available from teeth or skeletal parts. The development of molecular methods for age estimation allowing to use human specimens that possess no morphological age information, such as bloodstains, is extremely valuable as this type of samples is commonly found at crime scenes. Recently, we introduced a DNA-based approach for human age estimation from blood based on the quantification of T-cell specific DNA rearrangements (sjTRECs), which achieves accurate assignment of blood DNA samples to one of four 20-year-interval age categories. Aiming at improving the accuracy of molecular age estimation from blood, we investigated different types of biomarkers. We started out by systematic genome-wide surveys for new age-informative mRNA and DNA methylation markers in blood from the same young and old individuals using microarray technologies. The obtained candidate markers were validated in independent samples covering a wide age range using alternative technologies together with previously proposed DNA methylation, sjTREC, and telomere length markers. Cross-validated multiple regression analysis was applied for estimating and validating the age predictive power of various sets of biomarkers within and across different marker types. We found that DNA methylation markers outperformed mRNA, sjTREC, and telomere length in age predictive power. The best performing model included 8 DNA methylation markers derived from 3 CpG islands reaching a high level of accuracy (cross-validated R(2)=0.88, SE+/-6.97 years, mean absolute deviation 5.07 years). However, our data also suggest that mRNA markers can provide independent age information: a model using a combined set of 5 DNA methylation markers and one mRNA marker could provide similarly high accuracy (cross-validated R(2)=0.86, SE+/-7.62 years, mean absolute deviation 4.60 years). Overall, our study provides new and confirms previously suggested molecular biomarkers for age estimation from blood. Moreover, our comparative study design revealed that DNA methylation markers are superior for this purpose over other types of molecular biomarkers tested. While the new and some previous findings are highly promising, before molecular age estimation can eventually meet forensic practice, the proposed biomarkers should be tested further in larger sets of blood samples from both healthy and unhealthy individuals, and markers and genotyping methods shall be validated to meet forensic standards.

2015
- SkinResTechnolValidation of image analysis techniques to measure skin aging features from facial photographsM. A. Hamer, L. C. Jacobs, J. S. Lall, and 9 more authorsSkin Res Technol, 2015
BACKGROUND: Accurate measurement of the extent skin has aged is crucial for skin aging research. Image analysis offers a quick and consistent approach for quantifying skin aging features from photographs, but is prone to technical bias and requires proper validation. METHODS: Facial photographs of 75 male and 75 female North-European participants, randomly selected from the Rotterdam Study, were graded by two physicians using photonumeric scales for wrinkles (full face, forehead, crow’s feet, nasolabial fold and upper lip), pigmented spots and telangiectasia. Image analysis measurements of the same features were optimized using photonumeric grades from 50 participants, then compared to photonumeric grading in the 100 remaining participants stratified by sex. RESULTS: The inter-rater reliability of the photonumeric grades was good to excellent (intraclass correlation coefficients 0.65-0.93). Correlations between the digital measures and the photonumeric grading were moderate to excellent for all the wrinkle comparisons (Spearman’s rho rho = 0.52-0.89) bar the upper lip wrinkles in the men (fair, rho = 0.30). Correlations were moderate to good for pigmented spots and telangiectasia (rho = 0.60-0.75). CONCLUSION: These comparisons demonstrate that all the image analysis measures, bar the upper lip measure in the men, are suitable for use in skin aging research and highlight areas of improvement for future refinements of the techniques.
- JIDA Genome-Wide Association Study Identifies the Skin Color Genes IRF4, MC1R, ASIP, and BNC2 Influencing Facial Pigmented SpotsL. C. Jacobs, M. A. Hamer, D. A. Gunn, and 12 more authorsJ Invest Dermatol, 2015
Facial pigmented spots are a common skin aging feature, but genetic predisposition has yet to be thoroughly investigated. We conducted a genome-wide association study for pigmented spots in 2,844 Dutch Europeans from the Rotterdam Study (mean age: 66.9+/-8.0 years; 47% male). Using semi-automated image analysis of high-resolution digital facial photographs, facial pigmented spots were quantified as the percentage of affected skin area (mean women: 2.0% +/-0.9, men: 0.9% +/-0.6). We identified genome-wide significant association with pigmented spots at three genetic loci: IRF4 (rs12203592, P=1.8 x 10(-27)), MC1R (compound heterozygosity score, P=2.3 x 10(-24)), and RALY/ASIP (rs6059655, P=1.9 x 10(-9)). In addition, after adjustment for the other three top-associated loci the BNC2 locus demonstrated significant association (rs62543565, P=2.3 x 10(-8)). The association signals observed at all four loci were successfully replicated (P<0.05) in an independent Dutch cohort (Leiden Longevity Study n=599). Although the four genes have previously been associated with skin color variation and skin cancer risk, all association signals remained highly significant (P<2 x 10(-8)) when conditioning the association analyses on skin color. We conclude that genetic variations in IRF4, MC1R, RALY/ASIP, and BNC2 contribute to the acquired amount of facial pigmented spots during aging, through pathways independent of the basal melanin production.
- HumMolGenetIRF4, MC1R and TYR genes are risk factors for actinic keratosis independent of skin colorL. C. Jacobs, F. Liu, L. M. Pardo, and 4 more authorsHum Mol Genet, 2015
Actinic keratosis (AK) is a pre-malignant skin disease, highly prevalent in elderly Europeans. This study investigates genetic susceptibility to AK with a genome-wide association study (GWAS). A full body skin examination was performed in 3194 elderly individuals from the Rotterdam Study (RS) of exclusive north-western European origin (aged 51-99 years, 45% male). Physicians graded the number of AK into four severity levels: none (76%), 1-3 (14%), 4-9 (6%) and >/=10 (5%), and skin color was quantified using a spectrophotometer on sun-unexposed skin. A GWAS for AK severity was conducted, where promising signals at IRF4 and MC1R (P < 4.2 x 10(-7)) were successfully replicated in an additional cohort of 623 RS individuals (IRF4, rs12203592, Pcombined = 6.5 x 10(-13) and MC1R, rs139810560, Pcombined = 4.1 x 10(-9)). Further, in an analysis of ten additional well-known human pigmentation genes, TYR also showed significant association with AK (rs1393350, P = 5.3 x 10(-4)) after correction for multiple testing. Interestingly, the strength and significance of above-mentioned associations retained largely the same level after skin color adjustment. Overall, our data strongly suggest that IRF4, MC1R and TYR genes likely have pleiotropic effects, a combination of pigmentation and oncogenic functions, resulting in an increased risk of AK.
- HumGenetGenetics of skin color variation in Europeans: genome-wide association studies with functional follow-upF. Liu, M. Visser, D. L. Duffy, and 23 more authorsHum Genet, 2015
In the International Visible Trait Genetics (VisiGen) Consortium, we investigated the genetics of human skin color by combining a series of genome-wide association studies (GWAS) in a total of 17,262 Europeans with functional follow-up of discovered loci. Our GWAS provide the first genome-wide significant evidence for chromosome 20q11.22 harboring the ASIP gene being explicitly associated with skin color in Europeans. In addition, genomic loci at 5p13.2 (SLC45A2), 6p25.3 (IRF4), 15q13.1 (HERC2/OCA2), and 16q24.3 (MC1R) were confirmed to be involved in skin coloration in Europeans. In follow-up gene expression and regulation studies of 22 genes in 20q11.22, we highlighted two novel genes EIF2S2 and GSS, serving as competing functional candidates in this region and providing future research lines. A genetically inferred skin color score obtained from the 9 top-associated SNPs from 9 genes in 940 worldwide samples (HGDP-CEPH) showed a clear gradual pattern in Western Eurasians similar to the distribution of physical skin color, suggesting the used 9 SNPs as suitable markers for DNA prediction of skin color in Europeans and neighboring populations, relevant in future forensic and anthropological investigations.
- IntJCancerMC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP projectE. Pasquali, J. C. Garcia-Borron, M. C. Fargnoli, and 29 more authorsInt J Cancer, 2015
The MC1R gene is a key regulator of skin pigmentation. We aimed to evaluate the association between MC1R variants and the risk of sporadic cutaneous melanoma (CM) within the M-SKIP project, an international pooled-analysis on MC1R, skin cancer and phenotypic characteristics. Data included 5,160 cases and 12,119 controls from 17 studies. We calculated a summary odds ratio (SOR) for the association of each of the nine most studied MC1R variants and of variants combined with CM by using random-effects models. Stratified analysis by phenotypic characteristics were also performed. Melanoma risk increased with presence of any of the main MC1R variants: the SOR for each variant ranged from 1.47 (95%CI: 1.17-1.84) for V60L to 2.74 (1.53-4.89) for D84E. Carriers of any MC1R variant had a 66% higher risk of developing melanoma compared with wild-type subjects (SOR; 95%CI: 1.66; 1.41-1.96) and the risk attributable to MC1R variants was 28%. When taking into account phenotypic characteristics, we found that MC1R-associated melanoma risk increased only for darker-pigmented Caucasians: SOR (95%CI) was 3.14 (2.06-4.80) for subjects with no freckles, no red hair and skin Type III/IV. Our study documents the important role of all the main MC1R variants in sporadic CM and suggests that they have a direct effect on melanoma risk, independently on the phenotypic characteristics of carriers. This is of particular importance for assessing preventive strategies, which may be directed to darker-pigmented Caucasians with MC1R variants as well as to lightly pigmented, fair-skinned subjects.
- BrJCancerMC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP projectE. Tagliabue, M. C. Fargnoli, S. Gandini, and 21 more authorsBr J Cancer, 2015
BACKGROUND: The melanocortin-1-receptor (MC1R) gene regulates human pigmentation and is highly polymorphic in populations of European origins. The aims of this study were to evaluate the association between MC1R variants and the risk of non-melanoma skin cancer (NMSC), and to investigate whether risk estimates differed by phenotypic characteristics. METHODS: Data on 3527 NMSC cases and 9391 controls were gathered through the M-SKIP Project, an international pooled-analysis on MC1R, skin cancer and phenotypic characteristics. We calculated summary odds ratios (SOR) with random-effect models, and performed stratified analyses. RESULTS: Subjects carrying at least one MC1R variant had an increased risk of NMSC overall, basal cell carcinoma (BCC) and squamous cell carcinoma (SCC): SOR (95%CI) were 1.48 (1.24-1.76), 1.39 (1.15-1.69) and 1.61 (1.35-1.91), respectively. All of the investigated variants showed positive associations with NMSC, with consistent significant results obtained for V60L, D84E, V92M, R151C, R160W, R163Q and D294H: SOR (95%CI) ranged from 1.42 (1.19-1.70) for V60L to 2.66 (1.06-6.65) for D84E variant. In stratified analysis, there was no consistent pattern of association between MC1R and NMSC by skin type, but we consistently observed higher SORs for subjects without red hair. CONCLUSIONS: Our pooled-analysis highlighted a role of MC1R variants in NMSC development and suggested an effect modification by red hair colour phenotype.
2014
- JAMADermatolIntrinsic and extrinsic risk factors for sagging eyelidsL. C. Jacobs, F. Liu, I. Bleyen, and 9 more authorsJAMA Dermatol, 2014
IMPORTANCE: Sagging eyelids, or dermatochalasis, are a frequent concern in older adults. It is considered a feature of skin aging, but risk factors other than aging are largely unknown. OBJECTIVE: To study nongenetic and genetic risk factors for sagging eyelids. DESIGN: Upper eyelid sagging was graded in 4 categories of severity using digital photographs. Dermatochalasis was defined as the eyelid hanging over the eyelashes. Age, sex, skin color, tanning ability, hormonal status in women, current smoking, body mass index, and sun protection behavior were analyzed in a multivariable multinomial logistic regression model. Genetic predisposition was assessed using heritability analysis and a genome-wide association study. SETTING AND PARTICIPANTS: The study was performed in 2 independent population-based cohorts. The Rotterdam Study included older adults from one district in Rotterdam, the Netherlands, and the UK Adult Twin Registry (TwinsUK) included twins from all over the United Kingdom. Participants were 5578 unrelated Dutch Europeans (mean age, 67.1 years; 44.0% male) from the Rotterdam Study and 2186 twins (mean age, 53.1 years; 10.4% male) from the TwinsUK. MAIN OUTCOMES AND MEASURES: Sagging eyelid severity levels, ranging from 1 (normal control) to 4 (severe sagging). RESULTS: Among 5578 individuals from the Rotterdam Study, 17.8% showed dermatochalasis (moderate and severe sagging eyelids). Significant and independent risk factors for sagging eyelids included age, male sex, lighter skin color, and higher body mass index. In addition, current smoking was borderline significantly associated. Heritability of sagging eyelids was estimated to be 61% among 1052 twin pairs from the TwinsUK (15.6% showed dermatochalasis). A meta-analysis of genome-wide association study results from 5578 Rotterdam Study and 1053 TwinsUK participants showed a genome-wide significant recessive protective effect of the C allele of rs11876749 (P = 1.7 x 10(-8)). This variant is located close to TGIF1 (an inducer of transforming growth factor beta), which is a known gene associated with skin aging. CONCLUSIONS AND RELEVANCE: This is the first observational study to date demonstrating that other risk factors (male sex, genetic variants, lighter skin color, high body mass index, and possibly current smoking) in addition to aging are involved in the origin of sagging eyelids.
- PLoSComputBiolGAGA: a new algorithm for genomic inference of geographic ancestry reveals fine level population substructure in EuropeansO. Lao, F. Liu, A. Wollstein, and 1 more authorPLoS Comput Biol, 2014
Attempts to detect genetic population substructure in humans are troubled by the fact that the vast majority of the total amount of observed genetic variation is present within populations rather than between populations. Here we introduce a new algorithm for transforming a genetic distance matrix that reduces the within-population variation considerably. Extensive computer simulations revealed that the transformed matrix captured the genetic population differentiation better than the original one which was based on the T1 statistic. In an empirical genomic data set comprising 2,457 individuals from 23 different European subpopulations, the proportion of individuals that were determined as a genetic neighbour to another individual from the same sampling location increased from 25% with the original matrix to 52% with the transformed matrix. Similarly, the percentage of genetic variation explained between populations by means of Analysis of Molecular Variance (AMOVA) increased from 1.62% to 7.98%. Furthermore, the first two dimensions of a classical multidimensional scaling (MDS) using the transformed matrix explained 15% of the variance, compared to 0.7% obtained with the original matrix. Application of MDS with Mclust, SPA with Mclust, and GemTools algorithms to the same dataset also showed that the transformed matrix gave a better association of the genetic clusters with the sampling locations, and particularly so when it was used in the AMOVA framework with a genetic algorithm. Overall, the new matrix transformation introduced here substantially reduces the within population genetic differentiation, and can be broadly applied to methods such as AMOVA to enhance their sensitivity to reveal population substructure. We herewith provide a publically available (http://www.erasmusmc.nl/fmb/resources/GAGA) model-free method for improved genetic population substructure detection that can be applied to human as well as any other species data in future studies relevant to evolutionary biology, behavioural ecology, medicine, and forensics.
- IJLMPHOX2B polyalanine repeat length is associated with sudden infant death syndrome and unclassified sudden infant death in the Dutch populationG. Liebrechts-Akkerman, F. Liu, O. Lao, and 7 more authorsInt J Legal Med, 2014
Unclassified sudden infant death (USID) is the sudden and unexpected death of an infant that remains unexplained after thorough case investigation including performance of a complete autopsy and review of the circumstances of death and the clinical history. When the infant is below 1 year of age and with onset of the fatal episode apparently occurring during sleep, this is referred to as sudden infant death syndrome (SIDS). USID and SIDS remain poorly understood despite the identification of several environmental and some genetic risk factors. In this study, we investigated genetic risk factors involved in the autonomous nervous system in 195 Dutch USID/SIDS cases and 846 Dutch, age-matched healthy controls. Twenty-five DNA variants from 11 genes previously implicated in the serotonin household or in the congenital central hypoventilation syndrome, of which some have been associated with SIDS before, were tested. Of all DNA variants considered, only the length variation of the polyalanine repeat in exon 3 of the PHOX2B gene was found to be statistically significantly associated with USID/SIDS in the Dutch population after multiple test correction. Interestingly, our data suggest that contraction of the PHOX2B exon 3 polyalanine repeat that we found in six of 160 SIDS and USID cases and in six of 814 controls serves as a probable genetic risk factor for USID/SIDS at least in the Dutch population. Future studies are needed to confirm this finding and to understand the functional effect of the polyalanine repeat length variation, in particular contraction, in exon 3 of the PHOX2B gene.
- HumGenetCommon DNA variants predict tall stature in EuropeansF. Liu, A. E. Hendriks, A. Ralf, and 10 more authorsHum Genet, 2014
Genomic prediction of the extreme forms of adult body height or stature is of practical relevance in several areas such as pediatric endocrinology and forensic investigations. Here, we examine 770 extremely tall cases and 9,591 normal height controls in a population-based Dutch European sample to evaluate the capability of known height-associated DNA variants in predicting tall stature. Among the 180 normal height-associated single nucleotide polymorphisms (SNPs) previously reported by the Genetic Investigation of ANthropocentric Traits (GIANT) genome-wide association study on normal stature, in our data 166 (92.2 %) showed directionally consistent effects and 75 (41.7 %) showed nominally significant association with tall stature, indicating that the 180 GIANT SNPs are informative for tall stature in our Dutch sample. A prediction analysis based on the weighted allele sums method demonstrated a substantially improved potential for predicting tall stature (AUC = 0.75; 95 % CI 0.72-0.79) compared to a previous attempt using 54 height-associated SNPs (AUC = 0.65). The achieved accuracy is approaching practical relevance such as in pediatrics and forensics. Furthermore, a reanalysis of all SNPs at the 180 GIANT loci in our data identified novel secondary association signals for extreme tall stature at TGFB2 (P = 1.8 x 10(-13)) and PCSK5 (P = 7.8 x 10(-11)) suggesting the existence of allelic heterogeneity and underlining the importance of fine analysis of already discovered loci. Extrapolating from our results suggests that the genomic prediction of at least the extreme forms of common complex traits in humans including common diseases are likely to be informative if large numbers of trait-associated common DNA variants are available.
- FSIGThe common occurrence of epistasis in the determination of human pigmentation and its impact on DNA-based pigmentation phenotype predictionE. Pospiech, A. Wojas-Pelc, S. Walsh, and 6 more authorsForensic Sci Int Genet, 2014
The role of epistatic effects in the determination of complex traits is often underlined but its significance in the prediction of pigmentation phenotypes has not been evaluated so far. The prediction of pigmentation from genetic data can be useful in forensic science to describe the physical appearance of an unknown offender, victim, or missing person who cannot be identified via conventional DNA profiling. Available forensic DNA prediction systems enable the reliable prediction of several eye and hair colour categories. However, there is still space for improvement. Here we verified the association of 38 candidate DNA polymorphisms from 13 genes and explored the extent to which interactions between them may be involved in human pigmentation and their impact on forensic DNA prediction in particular. The model-building set included 718 Polish samples and the model-verification set included 307 independent Polish samples and additional 72 samples from Japan. In total, 29 significant SNP-SNP interactions were found with 5 of them showing an effect on phenotype prediction. For predicting green eye colour, interactions between HERC2 rs12913832 and OCA2 rs1800407 as well as TYRP1 rs1408799 raised the prediction accuracy expressed by AUC from 0.667 to 0.697 and increased the prediction sensitivity by >3%. Interaction between MC1R ’R’ variants and VDR rs731236 increased the sensitivity for light skin by >1% and by almost 3% for dark skin colour prediction. Interactions between VDR rs1544410 and TYR rs1042602 as well as between MC1R ’R’ variants and HERC2 rs12913832 provided an increase in red/non-red hair prediction accuracy from an AUC of 0.902-0.930. Our results thus underline epistasis as a common phenomenon in human pigmentation genetics and demonstrate that considering SNP-SNP interactions in forensic DNA phenotyping has little impact on eye, hair and skin colour prediction.
- FSIGDevelopmental validation of the HIrisPlex system: DNA-based eye and hair colour prediction for forensic and anthropological usageS. Walsh, L. Chaitanya, L. Clarisse, and 10 more authorsForensic Sci Int Genet, 2014
Forensic DNA Phenotyping or ’DNA intelligence’ tools are expected to aid police investigations and find unknown individuals by providing information on externally visible characteristics of unknown suspects, perpetrators and missing persons from biological samples. This is especially useful in cases where conventional DNA profiling or other means remain non-informative. Recently, we introduced the HIrisPlex system, capable of predicting both eye and hair colour from DNA. In the present developmental validation study, we demonstrate that the HIrisPlex assay performs in full agreement with the Scientific Working Group on DNA Analysis Methods (SWGDAM) guidelines providing an essential prerequisite for future HIrisPlex applications to forensic casework. The HIrisPlex assay produces complete profiles down to only 63 pg of DNA. Species testing revealed human specificity for a complete HIrisPlex profile, while only non-human primates showed the closest full profile at 20 out of the 24 DNA markers, in all animals tested. Rigorous testing of simulated forensic casework samples such as blood, semen, saliva stains, hairs with roots as well as extremely low quantity touch (trace) DNA samples, produced complete profiles in 88% of cases. Concordance testing performed between five independent forensic laboratories displayed consistent reproducible results on varying types of DNA samples. Due to its design, the assay caters for degraded samples, underlined here by results from artificially degraded DNA and from simulated casework samples of degraded DNA. This aspect was also demonstrated previously on DNA samples from human remains up to several hundreds of years old. With this paper, we also introduce enhanced eye and hair colour prediction models based on enlarged underlying databases of HIrisPlex genotypes and eye/hair colour phenotypes (eye colour: N = 9188 and hair colour: N = 1601). Furthermore, we present an online web-based system for individual eye and hair colour prediction from full and partial HIrisPlex DNA profiles. By demonstrating that the HIrisPlex assay is fully compatible with the SWGDAM guidelines, we provide the first forensically validated DNA test system for parallel eye and hair colour prediction now available to forensic laboratories for immediate casework application, including missing person cases. Given the robustness and sensitivity described here and in previous work, the HIrisPlex system is also suitable for analysing old and ancient DNA in anthropological and evolutionary studies.
2013
- HumGenetComprehensive candidate gene study highlights UGT1A and BNC2 as new genes determining continuous skin color variation in EuropeansL. C. Jacobs, A. Wollstein, O. Lao, and 6 more authorsHum Genet, 2013
Natural variation in human skin pigmentation is primarily due to genetic causes rooted in recent evolutionary history. Genetic variants associated with human skin pigmentation confer risk of skin cancer and may provide useful information in forensic investigations. Almost all previous gene-mapping studies of human skin pigmentation were based on categorical skin color information known to oversimplify the continuous nature of human skin coloration. We digitally quantified skin color into hue and saturation dimensions for 5,860 Dutch Europeans based on high-resolution skin photographs. We then tested an extensive list of 14,185 single nucleotide polymorphisms in 281 candidate genes potentially involved in human skin pigmentation for association with quantitative skin color phenotypes. Confirmatory association was revealed for several known skin color genes including HERC2, MC1R, IRF4, TYR, OCA2, and ASIP. We identified two new skin color genes: genetic variants in UGT1A were significantly associated with hue and variants in BNC2 were significantly associated with saturation. Overall, digital quantification of human skin color allowed detecting new skin color genes. The variants identified in this study may also contribute to the risk of skin cancer. Our findings are also important for predicting skin color in forensic investigations.
- IJLMFirst all-in-one diagnostic tool for DNA intelligence: genome-wide inference of biogeographic ancestry, appearance, relatedness, and sex with the Identitas v1 Forensic ChipB. Keating, A. T. Bansal, S. Walsh, and 20 more authorsInt J Legal Med, 2013
When a forensic DNA sample cannot be associated directly with a previously genotyped reference sample by standard short tandem repeat profiling, the investigation required for identifying perpetrators, victims, or missing persons can be both costly and time consuming. Here, we describe the outcome of a collaborative study using the Identitas Version 1 (v1) Forensic Chip, the first commercially available all-in-one tool dedicated to the concept of developing intelligence leads based on DNA. The chip allows parallel interrogation of 201,173 genome-wide autosomal, X-chromosomal, Y-chromosomal, and mitochondrial single nucleotide polymorphisms for inference of biogeographic ancestry, appearance, relatedness, and sex. The first assessment of the chip’s performance was carried out on 3,196 blinded DNA samples of varying quantities and qualities, covering a wide range of biogeographic origin and eye/hair coloration as well as variation in relatedness and sex. Overall, 95 % of the samples (N = 3,034) passed quality checks with an overall genotype call rate >90 % on variable numbers of available recorded trait information. Predictions of sex, direct match, and first to third degree relatedness were highly accurate. Chip-based predictions of biparental continental ancestry were on average 94 % correct (further support provided by separately inferred patrilineal and matrilineal ancestry). Predictions of eye color were 85 % correct for brown and 70 % correct for blue eyes, and predictions of hair color were 72 % for brown, 63 % for blond, 58 % for black, and 48 % for red hair. From the 5 % of samples (N = 162) with <90 % call rate, 56 % yielded correct continental ancestry predictions while 7 % yielded sufficient genotypes to allow hair and eye color prediction. Our results demonstrate that the Identitas v1 Forensic Chip holds great promise for a wide range of applications including criminal investigations, missing person investigations, and for national security purposes.
- SCDBColorful DNA polymorphisms in humansF. Liu, B. Wen, and M. KayserSemin Cell Dev Biol, 2013
In this review article we summarize current knowledge on how variation on the DNA level influences human pigmentation including color variation of iris, hair, and skin. We review recent progress in the field of human pigmentation genetics by focusing on the genes and DNA polymorphisms discovered to be involved in determining human pigmentation traits, their association with diseases particularly skin cancers, and their power to predict human eye, hair, and skin colors with potential utilization in forensic investigations.
- FSIGThe HIrisPlex system for simultaneous prediction of hair and eye colour from DNAS. Walsh, F. Liu, A. Wollstein, and 5 more authorsForensic Sci Int Genet, 2013
Recently, the field of predicting phenotypes of externally visible characteristics (EVCs) from DNA genotypes with the final aim of concentrating police investigations to find persons completely unknown to investigating authorities, also referred to as Forensic DNA Phenotyping (FDP), has started to become established in forensic biology. We previously developed and forensically validated the IrisPlex system for accurate prediction of blue and brown eye colour from DNA, and recently showed that all major hair colour categories are predictable from carefully selected DNA markers. Here, we introduce the newly developed HIrisPlex system, which is capable of simultaneously predicting both hair and eye colour from DNA. HIrisPlex consists of a single multiplex assay targeting 24 eye and hair colour predictive DNA variants including all 6 IrisPlex SNPs, as well as two prediction models, a newly developed model for hair colour categories and shade, and the previously developed IrisPlex model for eye colour. The HIrisPlex assay was designed to cope with low amounts of template DNA, as well as degraded DNA, and preliminary sensitivity testing revealed full DNA profiles down to 63pg input DNA. The power of the HIrisPlex system to predict hair colour was assessed in 1551 individuals from three different parts of Europe showing different hair colour frequencies. Using a 20% subset of individuals, while 80% were used for model building, the individual-based prediction accuracies employing a prediction-guided approach were 69.5% for blond, 78.5% for brown, 80% for red and 87.5% for black hair colour on average. Results from HIrisPlex analysis on worldwide DNA samples imply that HIrisPlex hair colour prediction is reliable independent of bio-geographic ancestry (similar to previous IrisPlex findings for eye colour). We furthermore demonstrate that it is possible to infer with a prediction accuracy of >86% if a brown-eyed, black-haired individual is of non-European (excluding regions nearby Europe) versus European (including nearby regions) bio-geographic origin solely from the strength of HIrisPlex eye and hair colour probabilities, which can provide extra intelligence for future forensic applications. The HIrisPlex system introduced here, including a single multiplex test assay, an interactive tool and prediction guide, and recommendations for reporting final outcomes, represents the first tool for simultaneously establishing categorical eye and hair colour of a person from DNA. The practical forensic application of the HIrisPlex system is expected to benefit cases where other avenues of investigation, including STR profiling, provide no leads on who the unknown crime scene sample donor or the unknown missing person might be.
2012
- PLoSGenetA genome-wide association study identifies five loci influencing facial morphology in EuropeansF. Liu, F. Lijn, C. Schurmann, and 31 more authorsPLoS Genet, 2012
Inter-individual variation in facial shape is one of the most noticeable phenotypes in humans, and it is clearly under genetic regulation; however, almost nothing is known about the genetic basis of normal human facial morphology. We therefore conducted a genome-wide association study for facial shape phenotypes in multiple discovery and replication cohorts, considering almost ten thousand individuals of European descent from several countries. Phenotyping of facial shape features was based on landmark data obtained from three-dimensional head magnetic resonance images (MRIs) and two-dimensional portrait images. We identified five independent genetic loci associated with different facial phenotypes, suggesting the involvement of five candidate genes–PRDM16, PAX3, TP63, C5orf50, and COL17A1–in the determination of the human face. Three of them have been implicated previously in vertebrate craniofacial development and disease, and the remaining two genes potentially represent novel players in the molecular networks governing facial development. Our finding at PAX3 influencing the position of the nasion replicates a recent GWAS of facial features. In addition to the reported GWA findings, we established links between common DNA variants previously associated with NSCL/P at 2p21, 8q24, 13q31, and 17q22 and normal facial-shape variations based on a candidate gene approach. Overall our study implies that DNA variants in genes essential for craniofacial development contribute with relatively small effect size to the spectrum of normal variation in human facial morphology. This observation has important consequences for future studies aiming to identify more genes involved in the human facial morphology, as well as for potential applications of DNA prediction of facial shape such as in future forensic applications.
- BMC-MRMMelanocortin-1 receptor, skin cancer and phenotypic characteristics (M-SKIP) project: study design and methods for pooling results of genetic epidemiological studiesS. Raimondi, S. Gandini, M. C. Fargnoli, and 35 more authorsBMC Med Res Methodol, 2012
BACKGROUND: For complex diseases like cancer, pooled-analysis of individual data represents a powerful tool to investigate the joint contribution of genetic, phenotypic and environmental factors to the development of a disease. Pooled-analysis of epidemiological studies has many advantages over meta-analysis, and preliminary results may be obtained faster and with lower costs than with prospective consortia. DESIGN AND METHODS: Based on our experience with the study design of the Melanocortin-1 receptor (MC1R) gene, SKin cancer and Phenotypic characteristics (M-SKIP) project, we describe the most important steps in planning and conducting a pooled-analysis of genetic epidemiological studies. We then present the statistical analysis plan that we are going to apply, giving particular attention to methods of analysis recently proposed to account for between-study heterogeneity and to explore the joint contribution of genetic, phenotypic and environmental factors in the development of a disease. Within the M-SKIP project, data on 10,959 skin cancer cases and 14,785 controls from 31 international investigators were checked for quality and recoded for standardization. We first proposed to fit the aggregated data with random-effects logistic regression models. However, for the M-SKIP project, a two-stage analysis will be preferred to overcome the problem regarding the availability of different study covariates. The joint contribution of MC1R variants and phenotypic characteristics to skin cancer development will be studied via logic regression modeling. DISCUSSION: Methodological guidelines to correctly design and conduct pooled-analyses are needed to facilitate application of such methods, thus providing a better summary of the actual findings on specific fields.
- FSIGDNA-based eye colour prediction across Europe with the IrisPlex systemS. Walsh, A. Wollstein, F. Liu, and 11 more authorsForensic Sci Int Genet, 2012
The ability to predict Externally Visible Characteristics (EVCs) from DNA, also referred to as Forensic DNA Phenotyping (FDP), is an exciting new chapter in forensic genetics holding great promise for tracing unknown individuals who are unidentifiable via standard forensic short tandem repeat (STR) profiling. For the purpose of DNA-based eye colour prediction, we previously developed the IrisPlex system consisting of a multiplex genotyping assay and a prediction model based on genotype and phenotype data from 3804 Dutch Europeans. Recently, we performed a forensic developmental validation study of the highly sensitive IrisPlex assay, which currently represents the only validated tool available for DNA-based prediction of eye colour in forensic applications. In the present study, we validate the IrisPlex prediction model by extending our initially described model towards genotype and phenotype data from multiple European populations. We performed IrisPlex analysis on 3840 individuals from seven sites across Europe as part of the European Eye (EUREYE) study for which DNA and high-resolution eye images were available. The accuracy rate of correctly predicting an individual’s eye colour as being blue or brown, above the empirically established probability threshold of 0.7, was on average 94% across all seven European populations, ranging from 91% to 98%, despite the large variation in eye colour frequencies between the populations. The overall prediction accuracies expressed by the area under the receiver characteristic operating curves (AUC) were 0.96 for blue and 0.96 for brown eyes, which is considerably higher than those established before. The IrisPlex prediction model parameters generated from this multi-population European dataset, and thus its prediction capabilities, were highly comparable to those previously established. Therefore, the increased information regarding eye colour phenotype and genotype distributions across Europe, and the system’s ability to provide eye colour predictions across Europe accurately, both highlight additional evidence for the utility of the IrisPlex system in forensic casework.
2011
- EJHGGenetic determination of human facial morphology: links between cleft-lips and normal variationS. Boehringer, F. Lijn, F. Liu, and 17 more authorsEur J Hum Genet, 2011
Recent genome-wide association studies have identified single nucleotide polymorphisms (SNPs) associated with non-syndromic cleft lip with or without cleft palate (NSCL/P), and other previous studies showed distinctly differing facial distance measurements when comparing unaffected relatives of NSCL/P patients with normal controls. Here, we test the hypothesis that genetic loci involved in NSCL/P also influence normal variation in facial morphology. We tested 11 SNPs from 10 genomic regions previously showing replicated evidence of association with NSCL/P for association with normal variation of nose width and bizygomatic distance in two cohorts from Germany (N=529) and the Netherlands (N=2497). The two most significant associations found were between nose width and SNP rs1258763 near the GREM1 gene in the German cohort (P=6 x 10(-4)), and between bizygomatic distance and SNP rs987525 at 8q24.21 near the CCDC26 gene (P=0.017) in the Dutch sample. A genetic prediction model explained 2% of phenotype variation in nose width in the German and 0.5% of bizygomatic distance variation in the Dutch cohort. Although preliminary, our data provide a first link between genetic loci involved in a pathological facial trait such as NSCL/P and variation of normal facial morphology. Moreover, we present a first approach for understanding the genetic basis of human facial appearance, a highly intriguing trait with implications on clinical practice, clinical genetics, forensic intelligence, social interactions and personal identity.
- HumGenetModel-based prediction of human hair color using DNA variantsW. Branicki, F. Liu, K. Duijn, and 6 more authorsHum Genet, 2011
Predicting complex human phenotypes from genotypes is the central concept of widely advocated personalized medicine, but so far has rarely led to high accuracies limiting practical applications. One notable exception, although less relevant for medical but important for forensic purposes, is human eye color, for which it has been recently demonstrated that highly accurate prediction is feasible from a small number of DNA variants. Here, we demonstrate that human hair color is predictable from DNA variants with similarly high accuracies. We analyzed in Polish Europeans with single-observer hair color grading 45 single nucleotide polymorphisms (SNPs) from 12 genes previously associated with human hair color variation. We found that a model based on a subset of 13 single or compound genetic markers from 11 genes predicted red hair color with over 0.9, black hair color with almost 0.9, as well as blond, and brown hair color with over 0.8 prevalence-adjusted accuracy expressed by the area under the receiver characteristic operating curves (AUC). The identified genetic predictors also differentiate reasonably well between similar hair colors, such as between red and blond-red, as well as between blond and dark-blond, highlighting the value of the identified DNA variants for accurate hair color prediction.
- JADAssociation of HSP70 and its co-chaperones with Alzheimer’s diseaseL. Broer, M. A. Ikram, M. Schuur, and 14 more authorsJ Alzheimers Dis, 2011
The heat shock protein (HSP) 70 family has been implicated in the pathology of Alzheimer’s disease (AD). In this study, we examined common genetic variations in the 80 genes encoding HSP70 and its co-chaperones. We conducted a study in a series of 462 patients and 5238 unaffected participants derived from the Rotterdam Study, a population-based study including 7983 persons aged 55 years and older. We genotyped a total of 12,053 Single Nucleotide Polymorphisms (SNPs) using the HumanHap550K Genotyping BeadChip from Illumina. Replication was performed in two independent cohort studies, the Framingham Heart study (FHS; n = 806) and Cardiovascular Health Study (CHS; n = 2150). When adjusting for multiple testing, we found a small but consistent, though not significant effect of rs12118313 located 32 kb from PFDN2, with an OR of 1.19 (p-value from meta-analysis = 0.003). However this SNP was in the intron of another gene, suggesting it is unlikely this SNP reflects the effect of PFDN2. In a formal pathway analysis we found nominally significant evidence for an association of BAG, DNAJA and prefoldin with AD. These findings corroborate with those of a study of 2032 AD patients and 5328 controls, in which several members of the prefoldin family showed evidence for association to AD. Our study did not reveal evidence for a genetic variant if the HSP70 family with a major effect on AD. However, our findings of the single SNP analysis and pathway analysis suggest that multiple genetic variants in prefoldin are associated with AD.
- EurJPediatrPostnatal parental smoking: an important risk factor for SIDSG. Liebrechts-Akkerman, O. Lao, F. Liu, and 5 more authorsEur J Pediatr, 2011
BACKGROUND: Sudden infant death syndrome (SIDS) is the unexpected death of an infant that remains unexplained after a thorough investigation of the circumstances, family history, paediatric investigation and complete autopsy. In Western society, it is the leading cause of post-neonatal death below 1 year of age. In the Netherlands, the SIDS incidence is very low, which offers opportunities to assess the importance of old and new environmental risk factors. For this purpose, cases were collected through pathology departments and the working group on SIDS of the Dutch Paediatrician Foundation. A total of 142 cases were included; these occurred after the parental education on sleeping position (1987), restricted to the international age criteria and had no histological explanation. Age-matched healthy controls (N = 2,841) came from a survey of the Netherlands Paediatric Surveillance Unit, completed between November 2002 and April 2003. A multivariate analysis was performed to determine the risk factors for SIDS, including sleeping position, antenatal maternal smoking, postnatal parental smoking, premature birth, gender, lack of breastfeeding and socio-economic status. Postnatal smoking was identified as an important environmental risk factor for SIDS (OR one parent = 2.5 [1.2, 5.0]; both parents = 5.77 [2.2, 15.5]; maternal = 2.7 [1.0, 6.4]; paternal = 2.4 [1.3, 4.5] ) as was prone sleeping (OR put prone to sleep = 21.5 [10.6, 43.5]; turned prone during sleep = 100 [46, 219]). Premature birth was also significantly associated with SIDS (OR = 2.4 [1.2, 4.8]). CONCLUSION: Postnatal parental smoking is currently a major environmental risk factor for SIDS in the Netherlands together with the long-established risk of prone sleeping.
- PLoSOneDetecting low frequent loss-of-function alleles in genome wide association studies with red hair color as exampleF. Liu, M. V. Struchalin, Kv Duijn, and 5 more authorsPLoS One, 2011
Multiple loss-of-function (LOF) alleles at the same gene may influence a phenotype not only in the homozygote state when alleles are considered individually, but also in the compound heterozygote (CH) state. Such LOF alleles typically have low frequencies and moderate to large effects. Detecting such variants is of interest to the genetics community, and relevant statistical methods for detecting and quantifying their effects are sorely needed. We present a collapsed double heterozygosity (CDH) test to detect the presence of multiple LOF alleles at a gene. When causal SNPs are available, which may be the case in next generation genome sequencing studies, this CDH test has overwhelmingly higher power than single SNP analysis. When causal SNPs are not directly available such as in current GWA settings, we show the CDH test has higher power than standard single SNP analysis if tagging SNPs are in linkage disequilibrium with the underlying causal SNPs to at least a moderate degree (r(2)>0.1). The test is implemented for genome-wide analysis in the publically available software package GenABEL which is based on a sliding window approach. We provide the proof of principle by conducting a genome-wide CDH analysis of red hair color, a trait known to be influenced by multiple loss-of-function alleles, in a total of 7,732 Dutch individuals with hair color ascertained. The association signals at the MC1R gene locus from CDH were uniformly more significant than traditional GWA analyses (the most significant P for CDH = 3.11x10(-)(1)(4)(2) vs. P for rs258322 = 1.33x10(-)(6)(6)). The CDH test will contribute towards finding rare LOF variants in GWAS and sequencing studies.
- JNNPGenetic risk factors for cerebral small-vessel disease in hypertensive patients from a genetically isolated populationM. Schuur, J. C. Swieten, S. Schol-Gelok, and 12 more authorsJ Neurol Neurosurg Psychiatry, 2011
BACKGROUND: Asymptomatic cerebral lesions on MRI such as white matter lesions (WML), lacunes and microbleeds are commonly seen in older people. We examined the role of a series of candidate genes involved in blood pressure regulation and amyloid metabolism. MATERIALS AND METHODS: The study was embedded in a family-based cohort sampled from a Dutch genetically isolated population. We selected individuals between 55 and 75 years of age with hypertension (N=129). Volumes of WML and presence of lacunes and microbleeds were assessed with MRI. We studied three genes involved in blood pressure regulation (angiotensin, angiotensin II type 1 receptor, alpha-adducin) and two genes involved in the amyloid pathway (apolipoprotein E (APOE) and sortilin-related receptor gene (SORL1)). RESULTS: All participants had WML (median volume, 3.1 ml; interquartile range, 1.5-6.5 ml); lacunar infarcts were present in 15.5% and microbleeds in 23.3%. Homozygosity for the APOE epsilon4 allele was associated with lacunes (OR, 4.8; 95% CI, 1.2 to 19.3). Individuals carrying two copies of the variant allele of four single nucleotide polymorphism (SNPs) located at the 3’-end of SORL1 (rs1699102, rs3824968, rs2282649, rs1010159) had significantly more often microbleeds (highest OR, 6.87; 95% CI, 1.78 to 26.44). CONCLUSION: The association of SORL1 with microbleeds suggests that the amyloid cascade is involved in the aetiology of microbleeds in populations with hypertension.
- FSIGIrisPlex: a sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry informationS. Walsh, F. Liu, K. N. Ballantyne, and 3 more authorsForensic Sci Int Genet, 2011
A new era of ’DNA intelligence’ is arriving in forensic biology, due to the impending ability to predict externally visible characteristics (EVCs) from biological material such as those found at crime scenes. EVC prediction from forensic samples, or from body parts, is expected to help concentrate police investigations towards finding unknown individuals, at times when conventional DNA profiling fails to provide informative leads. Here we present a robust and sensitive tool, termed IrisPlex, for the accurate prediction of blue and brown eye colour from DNA in future forensic applications. We used the six currently most eye colour-informative single nucleotide polymorphisms (SNPs) that previously revealed prevalence-adjusted prediction accuracies of over 90% for blue and brown eye colour in 6168 Dutch Europeans. The single multiplex assay, based on SNaPshot chemistry and capillary electrophoresis, both widely used in forensic laboratories, displays high levels of genotyping sensitivity with complete profiles generated from as little as 31pg of DNA, approximately six human diploid cell equivalents. We also present a prediction model to correctly classify an individual’s eye colour, via probability estimation solely based on DNA data, and illustrate the accuracy of the developed prediction test on 40 individuals from various geographic origins. Moreover, we obtained insights into the worldwide allele distribution of these six SNPs using the HGDP-CEPH samples of 51 populations. Eye colour prediction analyses from HGDP-CEPH samples provide evidence that the test and model presented here perform reliably without prior ancestry information, although future worldwide genotype and phenotype data shall confirm this notion. As our IrisPlex eye colour prediction test is capable of immediate implementation in forensic casework, it represents one of the first steps forward in the creation of a fully individualised EVC prediction system for future use in forensic DNA intelligence.
2010
- NeurobiolAgingThe apolipoprotein E gene and its age-specific effects on cognitive functionF. Liu, L. M. Pardo, M. Schuur, and 12 more authorsNeurobiol Aging, 2010
The E4 allele of the apolipoprotein E gene (APOE) is a well-established determinant of Alzheimer’s disease but its relation to cognitive function is much less understood. We studied the age-specific effects of the APOE*E4 allele on cognitive function and cardiovascular risk factors in 2208 related individuals. APOE*E4 allele was significantly associated with reduced test scores for Adult Verbal Learning Test, particularly on the memory and learning sub domains, in persons older than 50 years of age. The effect of APOE*E4 was independent of the effect of APOE*E4 on vascular risk factors and most pronounced on learning ability. Our findings suggest that APOE*E4 has an effect on cognitive function predominantly in the elderly, independent of vascular risk factors.
- PLoSGenetDigital quantification of human eye color highlights genetic association of three new lociF. Liu, A. Wollstein, P. G. Hysi, and 17 more authorsPLoS Genet, 2010
Previous studies have successfully identified genetic variants in several genes associated with human iris (eye) color; however, they all used simplified categorical trait information. Here, we quantified continuous eye color variation into hue and saturation values using high-resolution digital full-eye photographs and conducted a genome-wide association study on 5,951 Dutch Europeans from the Rotterdam Study. Three new regions, 1q42.3, 17q25.3, and 21q22.13, were highlighted meeting the criterion for genome-wide statistically significant association. The latter two loci were replicated in 2,261 individuals from the UK and in 1,282 from Australia. The LYST gene at 1q42.3 and the DSCR9 gene at 21q22.13 serve as promising functional candidates. A model for predicting quantitative eye colors explained over 50% of trait variance in the Rotterdam Study. Over all our data exemplify that fine phenotyping is a useful strategy for finding genes involved in human complex traits.
- BiolPsychiatryA genome-wide screen for depression in two independent Dutch populationsS. Schol-Gelok, A. C. Janssens, H. Tiemeier, and 10 more authorsBiol Psychiatry, 2010
BACKGROUND: Depression has a strong genetic component but candidate gene studies conducted to date have not shown consistent associations. METHODS: We conducted a genome-wide parametric and nonparametric linkage analysis in a large-scale family-based study including 115 individuals with depression who were identified based on the Hospital Anxiety Depression Scale, Center for Epidemiologic Studies Depression Rating Scale, or use of antidepressive medication. Further, we investigated the most promising chromosomal regions found in the genome-wide linkage analysis with an association analysis in 734 individuals in the family-based study and 2373 individuals in the population-based study. RESULTS: Our study demonstrated evidence for significant linkage of depression to chromosome 2p16.1-15 (logarithm of odds [LOD] = 5.13; parametric analysis) and suggestive evidence for linkage in nonparametric analysis to chromosome 5p15.33 (LOD = 2.14), 11q25 (LOD = 2.27), and 19p13.3 (LOD = 2.66). The subsequent association analysis in the family-based study showed region-wide significant association in intron 1 of the OPCML gene on chromosome 11q25 (empirical p value = .04). The association analysis in the population-based study did not show any region-wide significant association, yet showed suggestive association in intron 1 of the APLP2 gene on chromosome 11q25. CONCLUSIONS: Our linkage and association studies suggest a locus for depression on chromosomes 2p16.1-15 and 11q25. The linkage to chromosome 11q25 may be, in part, explained by the OPCML or the APLP2 gene. Further, there is evidence for a role of the GNG7 gene (chromosome 19p13.3).
2009
- HumMolGenetA genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variationK. Estrada, M. Krawczak, S. Schreiber, and 25 more authorsHum Mol Genet, 2009
Northwestern Europeans are among the tallest of human populations. The increase in body height in these people appears to have reached a plateau, suggesting the ubiquitous presence of an optimal environment in which genetic factors may have exerted a particularly strong influence on human growth. Therefore, we performed a genome-wide association study (GWAS) of body height using 2.2 million markers in 10 074 individuals from three Dutch and one German population-based cohorts. Upon genotyping, the 12 most significantly height-associated single nucleotide polymorphisms (SNPs) from this GWAS in 6912 additional individuals of Dutch and Swedish origin, a genetic variant (rs6717918) on chromosome 2q37.1 was found to be associated with height at a genome-wide significance level (P(combined) = 3.4 x 10(-9)). Notably, a second SNP (rs6718438) located approximately 450 bp away and in strong LD (r(2) = 0.77) with rs6717918 was previously found to be suggestive of a height association in 29 820 individuals of mainly northwestern European ancestry, and the over-expression of a nearby natriuretic peptide precursor type C (NPPC) gene, has been associated with overgrowth and skeletal anomalies. We also found a SNP (rs10472828) located on 5p14 near the natriuretic peptide receptor 3 (NPR3) gene, encoding a receptor of the NPPC ligand, to be associated with body height (P(combined) = 2.1 x 10(-7)). Taken together, these results suggest that variation in the C-type natriuretic peptide signaling pathway, involving the NPPC and NPR3 genes, plays an important role in determining human body height.
- Biol PsychiatryThe GAB2 gene and the risk of Alzheimer’s disease: replication and meta-analysisM. A. Ikram, F. Liu, B. A. Oostra, and 3 more authorsBiol Psychiatry, 2009
BACKGROUND: Recently, GAB2 has been suggested to modify the risk of late-onset Alzheimer’s disease (AD) among APOEepsilon4 carriers. However, replication data are inconsistent. METHODS: In a population-based cohort study (n = 5507; age > 55) with 443 incident AD cases, we investigated the association between rs4945261 and AD. Because we used high-density genotyping, we also investigated other polymorphisms within and around GAB2 and performed a meta-analysis with published studies. RESULTS: We found that rs4945261 was associated with AD among APOEepsilon4 carriers (p = .02) but not among noncarriers (p = .26). Fifteen of the 20 remaining polymorphisms within GAB2 and several polymorphisms in the 250kbp-region surrounding GAB2 were also associated with AD among carriers and only one among noncarriers. For rs2373115, meta-analysis yielded an odds ratio of 1.58 (1.17-2.14) with p = 3.0 * 10(-3) among carriers and 1.09 (.97-1.23) with p = .16 among noncarriers. For rs4945261, the pooled odds ratio was 1.75 (1.21-2.55) with p = 3.0 * 10(-3) among carriers and 1.20 (1.01-1.41) with p = .03 among noncarriers. CONCLUSIONS: We found GAB2 to be associated with AD. Furthermore, the meta-analysis also suggests that GAB2 modifies the risk of AD in APOEepsilon4 carriers.
- JADA study of the SORL1 gene in Alzheimer’s disease and cognitive functionF. Liu, M. A. Ikram, A. C. Janssens, and 15 more authorsJ Alzheimers Dis, 2009
Several studies have investigated the role of the neuronal sortilin-related receptor (SORL1) gene in Alzheimer’s disease (AD), but findings have been inconsistent. We conducted a study of 7 single nucleotide polymorphisms (SNPs), rs668387, rs689021, rs641120, rs1699102, rs3824968, rs2282649, and rs1010159, in the SORL1 gene that were associated to AD in previous studies. We tested for association with AD and cognitive function in 6741 participants of the Rotterdam Study and in 2883 individuals from the Erasmus Rucphen Family study. We performed meta-analyses on AD using our data together with those of previous studies published prior to September 2008 in Caucasians. Further, we studied up to 76 SNPs in a 400 kb region within and flanking the gene to evaluate the evidence that other genetic variants are associated with AD or cognitive function. There was no significant evidence for association between SORL1 SNPs and incident AD patients in the Rotterdam Study. In a meta-analysis of our data with those of others, six out of seven SNPs attained borderline significance. However, removal of the first study reporting association from the meta-analysis resulted in non-significant odds ratios for all SNPs. SNPs rs668387, rs689021, and rs641120 were associated with cognitive function in non-demented individuals at borderline statistical significance in two independent Dutch cohorts, but in the opposite direction. Testing for association using dense SNPs in the SORL1 gene did not reveal significant association with AD, or with cognitive function when adjusting for multiple testing. In conclusion, our data do not support the hypothesis that genetic variants in SORL1 are related to the risk of AD.
2008
- EJHGFamilial aggregation of preeclampsia and intrauterine growth restriction in a genetically isolated population in The NetherlandsA. L. Berends, E. A. Steegers, A. Isaacs, and 5 more authorsEur J Hum Genet, 2008
Preeclampsia and intrauterine growth restriction are related, pregnancy-specific disorders with a substantial genetic influence, which may have a joint genetic aetiology. We investigated familial aggregation, consanguinity and parent-of-origin effects for preeclampsia and IUGR. Fifty women with previous preeclampsia and 56 with previous pregnancies complicated by intrauterine growth restriction were recruited from a recent genetically isolated population in the Netherlands. Their relationships were estimated by means of a large genealogy database that contains information on more than 110 000 individuals from the isolate over 23 generations. Relationships were quantified using kinship and inbreeding coefficients. Parent-of-origin effects were evaluated by comparing parental kinships. Eighty-six women (39 preeclampsia and 47 intrauterine growth restriction) could be linked to one common ancestor within 14 generations. The proportion of related women with previous preeclampsia (95.6%) or pregnancies complicated by intrauterine growth restriction (95.1%) was significantly greater than expected by chance (P<0.001). Combined analysis of both disorders did not change the magnitude of familial aggregation. The proportion of women born from consanguineous marriages was increased in women with previous preeclampsia (81.8%) and those with intrauterine growth restriction (78%) compared to a random sample (P<0.001). Maternal and paternal kinships were not significantly different in both disorders. We demonstrate cosegregation of preeclampsia and intrauterine growth restriction, supporting a common genetic aetiology. The high proportion of parental consanguineous marriages suggests the possibility of an underlying recessive mutation. No evidence was found for a parent-of-origin effect either in preeclampsia or in intrauterine growth restriction.
- ArchNeurolMaternal transmission of multiple sclerosis in a dutch populationI. A. Hoppenbrouwers, F. Liu, Y. S. Aulchenko, and 4 more authorsArch Neurol, 2008
OBJECTIVE: To investigate the parental relationship of patients with multiple sclerosis (MS) from an extended pedigree with extensive genealogical information up to the middle of the 18th century. DESIGN: Multiple sclerosis is a complex disease resulting from genetic and environmental factors. Parent-of-origin effect, a phenomenon when the same allele may express differently depending on the sex of the transmitting parent, may influence the risk for MS. We investigated parental relationships between patients with MS using extensive genealogical information available from the Genetic Research in Isolated Populations program. We compared the average kinship of the parents of MS patients. We further explored the distribution of shortest genealogical links between parents of MS patients. Subjects Twenty-four MS patients from the isolated population who could be linked within a large complex pedigree, including 2471 people in total. RESULTS: The results consistently indicate a higher prevalence of maternal transmission of MS. The kinship between mothers of patients was 3.8 times higher than that between fathers (bootstrap P = .01). Among the 814 shortest connections between parents, 333 were maternal (40.9%, vs 25.0% expected), 98 were paternal (12.0%, vs 25.0% expected), and 383 were maternal-paternal (47.1%, vs 50.0% expected) (P < .001). CONCLUSIONS: Mothers of MS patients were more closely related than their fathers. This skewed relationship shows evidence for a maternal effect in MS. The most likely explanation is a gene-environment effect that takes place in utero.
- AJHGThree genome-wide association studies and a linkage analysis identify HERC2 as a human iris color geneM. Kayser, F. Liu, A. C. Janssens, and 19 more authorsAm J Hum Genet, 2008
Human iris color was one of the first traits for which Mendelian segregation was established. To date, the genetics of iris color is still not fully understood and is of interest, particularly in view of forensic applications. In three independent genome-wide association (GWA) studies of a total of 1406 persons and a genome-wide linkage study of 1292 relatives, all from the Netherlands, we found that the 15q13.1 region is the predominant region involved in human iris color. There were no other regions showing consistent genome-wide evidence for association and linkage to iris color. Single nucleotide polymorphisms (SNPs) in the HERC2 gene and, to a lesser extent, in the neighboring OCA2 gene were independently associated to iris color variation. OCA2 has been implicated in iris color previously. A replication study within two populations confirmed that the HERC2 gene is a new and significant determinant of human iris color variation, in addition to OCA2. Furthermore, HERC2 rs916977 showed a clinal allele distribution across 23 European populations, which was significantly correlated to iris color variation. We suggest that genetic variants regulating expression of the OCA2 gene exist in the HERC2 gene or, alternatively, within the 11.7 kb of sequence between OCA2 and HERC2, and that most iris color variation in Europeans is explained by those two genes. Testing markers in the HERC2-OCA2 region may be useful in forensic applications to predict eye color phenotypes of unknown persons of European genetic origin.
- EJHGAn approach for cutting large and complex pedigrees for linkage analysisF. Liu, A. Kirichenko, T. I. Axenovich, and 2 more authorsEur J Hum Genet, 2008
Utilizing large pedigrees in linkage analysis is a computationally challenging task. The pedigree size limits applicability of the Lander-Green-Kruglyak algorithm for linkage analysis. A common solution is to split large pedigrees into smaller computable subunits. We present a pedigree-splitting method that, within a user supplied bit-size limit, identifies subpedigrees having the maximal number of subjects of interest (eg patients) who share a common ancestor. We compare our method with the maximum clique partitioning method using a large and complex human pedigree consisting of 50 patients with Alzheimer’s disease ascertained from genetically isolated Dutch population. We show that under a bit-size limit our method can assign more patients to subpedigrees than the clique partitioning method, particularly when splitting deep pedigrees where the subjects of interest are scattered in recent generations and are relatively distantly related via multiple genealogic connections. Our pedigree-splitting algorithm and associated software can facilitate genome-wide linkage scans searching for rare mutations in large pedigrees coming from genetically isolated populations. The software package PedCut implementing our approach is available at http://mga.bionet.nsc.ru/soft/index.website.
- HumGenetThe MSX1 allele 4 homozygous child exposed to smoking at periconception is most sensitive in developing nonsyndromic orofacial cleftsM. J. Boogaard, D. Costa, I. P. Krapels, and 5 more authorsHum Genet, 2008
Nonsyndromic orofacial clefts (OFC) are common birth defects caused by certain genes interacting with environmental factors. Mutations and association studies indicate that the homeobox gene MSX1 plays a role in human clefting. In a Dutch case-control triad study (mother, father, and child), we investigated interactions between MSX1 and the parents’ periconceptional lifestyle in relation to the risk of OFC in their offspring. We studied 181 case- and 132 control mothers, 155 case- and 121 control fathers, and 176 case- and 146 control children, in which there were 107 case triads and 66 control triads. Univariable and multivariable logistic regression analyses were applied, and odds ratios (OR), 95% confidence intervals (CI) were calculated. Allele 4 of the CA marker in the MSX1 gene, consisting of nine CA repeats, was the most common allele found in both the case and control triads. Significant interactions were observed between allele 4 homozygosity of the child with maternal smoking (OR 2.7, 95% CI 1.1-6.6) and with smoking by both parents (OR 4.9, 95% CI 1.4-18.0). Allele 4 homozygosity in the mother and smoking showed a risk estimate of OR 3.2 (95% CI 1.1-9.0). If allele 4 homozygous mothers did not take daily folic acid supplements in the recommended periconceptional period, this also increased the risk of OFC for their offspring (OR 2.8, 95% CI 1.1-6.7). Our findings show that, in the Dutch population, periconceptional smoking by both parents interacts with a specific allelic variant of MSX1 to significantly increase OFC risk for their offspring. Possible underlying mechanisms are discussed.
2007
- NeurosciLettRelationship of the Ubiquilin 1 gene with Alzheimer’s and Parkinson’s disease and cognitive functionA. Arias-Vasquez, L. Lau, L. Pardo, and 9 more authorsNeurosci Lett, 2007
Ubiquilin 1 (UBQLN1) is involved in the ubiquitination machinery, which has been implicated in Alzheimer’s disease (AD) as well as Parkinson’s disease (PD). A polymorphism in the gene encoding for UBQLN1 has been previously associated with a higher risk of AD. We studied the role of the SNP rs12344615 on the UBQLN 1 gene in AD, PD and cognitive function in a population-based study, the Rotterdam Study, and a family-based study embedded in the genetic research in isolated population (GRIP) program. The Rotterdam Study includes 549 patients with AD and 157 patients with PD. The GRIP program includes a series of 123 patients with AD and a study of 1049 persons who are characterized for cognitive function. Data were analysed using logistic and multiple regression analysis. We found no significant difference in risk of AD or PD by the UBQLN1 SNP rs12344615 in our overall and stratified analyses in the Rotterdam Study. In our family-based study, we did not find evidence for linkage of AD to the region including the UBQLN1 gene. In the family-based study we also failed to detect an effect of this polymorphism on cognitive function. Our results suggest that it is unlikely that the SNP rs12344615 of the UBQLN1 gene is related to the onset of AD, PD or cognitive function.
- AJHGA genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch populationF. Liu, A. Arias-Vasquez, K. Sleegers, and 11 more authorsAm J Hum Genet, 2007
Alzheimer disease (AD) is the most common cause of dementia. We conducted a genome screen of 103 patients with late-onset AD who were ascertained as part of the Genetic Research in Isolated Populations (GRIP) program that is conducted in a recently isolated population from the southwestern area of The Netherlands. All patients and their 170 closely related relatives were genotyped using 402 microsatellite markers. Extensive genealogy information was collected, which resulted in an extremely large and complex pedigree of 4,645 members. The pedigree was split into 35 subpedigrees, to reduce the computational burden of linkage analysis. Simulations aiming to evaluate the effect of pedigree splitting on false-positive probabilities showed that a LOD score of 3.64 corresponds to 5% genomewide type I error. Multipoint analysis revealed four significant and one suggestive linkage peaks. The strongest evidence of linkage was found for chromosome 1q21 (heterogeneity LOD [HLOD]=5.20 at marker D1S498). Approximately 30 cM upstream of this locus, we found another peak at 1q25 (HLOD=4.0 at marker D1S218). These two loci are in a previously established linkage region. We also confirmed the AD locus at 10q22-24 (HLOD=4.15 at marker D10S185). There was significant evidence of linkage of AD to chromosome 3q22-24 (HLOD=4.44 at marker D3S1569). For chromosome 11q24-25, there was suggestive evidence of linkage (HLOD=3.29 at marker D11S1320). We next tested for association between cognitive function and 4,173 single-nucleotide polymorphisms in the linked regions in an independent sample consisting of 197 individuals from the GRIP region. After adjusting for multiple testing, we were able to detect significant associations for cognitive function in four of five AD-linked regions, including the new region on chromosome 3q22-24 and regions 1q25, 10q22-24, and 11q25. With use of cognitive function as an endophenotype of AD, our study indicates the that the RGSL2, RALGPS2, and C1orf49 genes are the potential disease-causing genes at 1q25. Our analysis of chromosome 10q22-24 points to the HTR7, MPHOSPH1, and CYP2C cluster. This is the first genomewide screen that showed significant linkage to chromosome 3q23 markers. For this region, our analysis identified the NMNAT3 and CLSTN2 genes. Our findings confirm linkage to chromosome 11q25. We were unable to confirm SORL1; instead, our analysis points to the OPCML and HNT genes.
2006
- AnnHumGenetIgnoring distant genealogic loops leads to false-positives in homozygosity mappingF. Liu, S. Elefante, C. M. Duijn, and 1 more authorAnn Hum Genet, 2006
Distant consanguineous loops are often unknown or ignored during homozygosity mapping analysis. This may potentially lead to an increased rate of false-positive linkage results. We show that failure to take into account the distant loops may seriously underestimate the degree of consanguinity, especially for people from genetically isolated populations; in 6 Alzheimer’s disease (AD) patients the distant loops accounted for 57.7 % of inbreeding on average. Theoretical evaluation showed that ignoring distant loops, which account for 18-75% of inbreeding, inflates the frequency of false positive conclusions substantially in 2-point linkage analysis, up to several hundred times. In multipoint linkage analysis of the 6 AD patients a chromosome-wide "empirical" significance of 5% corresponded to a true false positive rate of 11.1%. We show that converting multiple loops to a hypothetical loop capturing all inbreeding may be a convenient solution to avoid false positive results. When extended genealogic data are not available a hypothetical loop may still be constructed based on genomic data.